


Análise por Espectrometria de Fluorescência de Raios-X
Análise por espectrometria de fluorescência de raios-X
A fluorescência de raios X (XRF), é uma técnica espectroscópica de emissão que tem encontrado ampla aplicação na área de identificação e determinação elementar. A técnica depende da emissão de radiação x característica, normalmente na faixa de energia de 1 keV a 60 keV, após a excitação dos níveis de energia dos elétrons atômicos por uma fonte de energia externa, como um feixe de elétrons, um feixe de partículas carregadas ou um x feixe de raios. Na maioria das matrizes de amostras, a espectrometria de raios X pode detectar elementos em concentrações inferiores a 1 micrograma/g de amostra (1 ppm). Em uma amostra de filme fino, ele pode detectar quantidades totais de alguns décimos de um micrograma. Inicialmente, a espectrometria de raios X encontrou ampla aceitação em aplicações relacionadas a análises metalúrgicas e geoquímicas. Mais recentemente, a espectrometria de raios X provou ser valiosa na análise de amostras ambientais, na determinação de enxofre e elementos de desgaste em produtos petrolíferos, em aplicações envolvendo amostras forenses e em medições de materiais eletrônicos e informáticos.
A espectrometria de fluorescência de raios X (XRF) é uma ferramenta versátil em muitos problemas analíticos. Elementos maiores, menores e traços podem ser determinados qualitativa e quantitativamente em vários tipos de amostras, como metais, ligas, vidros, cimentos, minerais, rochas, minérios, polímeros, bem como materiais ambientais e biológicos. Elementos de sódio (Na) a urânio (U) são rotineiramente determinados usando o espectrômetro de fluorescência de raios-x com dispersão de energia (EDXRF), enquanto a aplicação do espectrômetro de fluorescência de raios-x com dispersão de comprimento de onda (WDXRF) permite a determinação eficiente de elementos de baixo Z até mesmo berílio (Be). Embora as amostras possam ser analisadas sem tratamento, resultados de alta qualidade podem ser garantidos se a preparação apropriada da amostra for aplicada. Isso pode variar desde a simples limpeza e polimento da amostra (metais, ligas), pulverização e peletização com ou sem ligante (cerâmica, minerais, minérios, solos, etc.), fusão da amostra com fluxo apropriado (cerâmica, rochas, minérios, etc.) à digestão com ácidos (metais, ligas). Desta forma, podem ser eliminados ou minimizados os erros resultantes da rugosidade da superfície, efeito do tamanho das partículas ou falta de homogeneidade do material.
Roentgen descobriu os raios X em 1895. H.G.J. Moseley desenvolveu as relações entre estrutura atômica e emissão de raios X e em 1913 publicou os primeiros espectros de raios X, que são a base da moderna espectrometria de raios X. Moseley reconheceu o potencial para determinações elementares quantitativas usando técnicas de raios-x. O desenvolvimento da instrumentação de raios X de rotina, levando ao espectrômetro de raios X conhecido hoje, ocorreu nas décadas seguintes. Coolidge projetou um tubo de raios X em 1913 que é semelhante aos usados atualmente. Soller alcançou a colimação de raios-x em 1924. Melhorias no detector de raios-x de gás por Geiger e Mueller em 1928 levaram ao projeto do primeiro WDXRF comercial por Friedman e Birks em 1948. Mais recentemente, outros detectores, como o germânio e os detectores semicondutores de silício dopado com lítio resultaram em projetos modificados de espectrômetro de raios-x. A instrumentação dispersiva de energia moderna facilita a identificação qualitativa de elementos em várias amostras. O conteúdo de informação de um espectro de raios X dispersivo de energia está entre os mais altos obtidos de materiais inorgânicos em uma única medição. A posição e a intensidade dos picos espectrais fornecem informações qualitativas e quantitativas, e a intensidade do fundo fornece informações sobre a composição da matriz da amostra.
A espectrometria de raios X é uma das poucas técnicas que podem ser aplicadas a amostras sólidas de diferentes formas. Embora a maioria dos espectrômetros de XRF sejam usados em laboratórios, muitos estão encontrando aplicação em análises de rotina para produção e controle de qualidade e em tarefas especializadas. Um diagrama estrutural do espectrômetro EDXRF é dado na Fig 1.
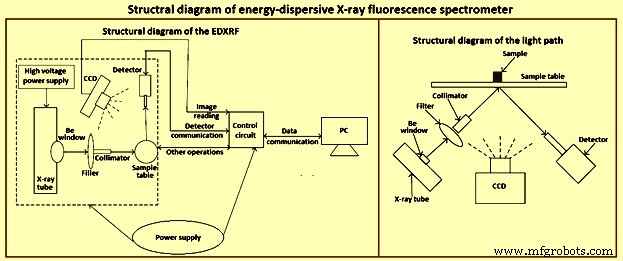
Fig 1 Diagrama estrutural do espectrômetro EDXRF
Radiação eletromagnética
A radiação eletromagnética é uma forma de energia que pode ser propagada através do espaço e pode interagir com átomos e moléculas para alterar seu estado de energia. Ambas as propriedades são importantes para a espectroscopia. A radiação eletromagnética mostra um comportamento que precisa de duas teorias para explicar. A teoria das ondas descreve o comportamento da radiação eletromagnética, como refração, reflexão, difração e dispersão. A radiação é definida como uma forma de energia que consiste em duas ondas ortogonais, cada uma com a mesma frequência e comprimento de onda. Um é um campo elétrico oscilante e o outro um campo magnético oscilante, produzindo assim o termo radiação eletromagnética.
No vácuo, a velocidade de propagação da onda através do espaço é a velocidade da luz (c =3 × 10 elevado à potência 10 cm/s). Isso leva a uma importante relação fundamental representada pela equação w.v =c. Esta expressão afirma que o produto do comprimento de onda (w) da radiação eletromagnética e sua frequência (v) é igual à sua velocidade. O comprimento de onda da radiação eletromagnética varia em muitas ordens de magnitude. Por exemplo, as ondas de rádio na faixa de transmissão AM normal têm comprimentos de onda de várias centenas de metros e comprimentos de onda ultravioleta estão na faixa de 10 nm a 100 nm (nanômetros). Em contraste, os raios X úteis na espectroscopia variam de 0,01 nm a 10 nm (Fig. 2).

Fig 2 Raios-X e outras radiações eletromagnéticas
Para espectrometria dispersiva de comprimento de onda, geralmente é mais conveniente usar unidades de comprimento de onda, mas para espectrometria de raios X com dispersão de energia (EDS), a descrição da energia é mais conveniente. No entanto, a interconversão é simples.
Várias descrições normalmente usadas das características dos raios X são significativas. O significado adequado da intensidade da radiação eletromagnética é a energia por unidade de área por unidade de tempo; no entanto, o número de contagens por unidade de tempo do detector é frequentemente usado como intensidade. Como a área é a área ativa do detector usado e o tempo é um parâmetro ajustável, o uso de contagens é uma descrição prática da intensidade dos raios X. Os termos raios X duros ou moles são frequentemente usados para diferenciar raios X de comprimentos de onda curtos (0,01 nm a 0,1 nm) e longos (0,1 nm a 1 nm), respectivamente. A radiação X cai na região de alta energia do espectro eletromagnético.
Emissão de raios-X
Os raios X são gerados a partir da perturbação dos orbitais de elétrons dos átomos. Isso pode ser feito de várias maneiras, sendo a mais comum o bombardeio de um elemento alvo com elétrons de alta energia, raios X ou partículas carregadas aceleradas. Os dois primeiros são frequentemente usados na espectrometria de raios X direta ou indiretamente. O bombardeio de elétrons resulta em um continuum de energias de raios X, bem como na radiação característica do elemento alvo. Ambos os tipos de radiação são encontrados na espectrometria de raios X.
Contínuo – A emissão de raios X com uma função suave e contínua de intensidade em relação à energia é chamada de radiação contínua ou bremsstrahlung. Um contínuo de raios X pode ser gerado de várias maneiras. No entanto, o mais útil é o feixe de elétrons usado para bombardear um alvo em um tubo de raios X. O contínuo é gerado como resultado da desaceleração progressiva de elétrons de alta energia colidindo com um alvo, que é uma distribuição de elétrons orbitais de várias energias. À medida que os elétrons que colidem interagem com os elétrons orbitais ligados, parte de sua energia cinética é convertida em radiação. A quantidade convertida depende da energia de ligação do elétron envolvido. Portanto, existe uma probabilidade estatística de quanta energia é convertida em cada interação.
A probabilidade de um elétron incidente interagir com um elétron orbital do elemento alvo aumenta com o número atômico do elemento, assim, a intensidade da emissão contínua aumenta com o número atômico do elemento alvo. Além disso, a probabilidade de uma interação aumenta com o número de elétrons por unidade de tempo no feixe, ou fluxo. Assim, a intensidade do contínuo aumenta com a corrente do feixe de elétrons, expressa em mili-ampères. Além disso, a capacidade dos elétrons colidentes de interagir com elétrons fortemente ligados do elemento alvo aumenta com a energia cinética dos elétrons bombardeadores. Como a energia cinética dos elétrons no feixe aumenta com o potencial de aceleração, a intensidade integrada do contínuo aumenta com o potencial de aceleração do elétron, expresso em quilovolts. Finalmente, a energia máxima manifestada como fótons de raios X é igual à energia cinética do elétron que colide, que por sua vez se relaciona com o potencial de aceleração. A energia da intensidade máxima no contínuo situa-se em cerca de dois terços da energia máxima emitida. Além disso, há a absorção de raios X dentro do material alvo ou absorção por materiais usados para janelas no tubo de raios X e detectores. Portanto, algumas modificações na distribuição de intensidade podem ocorrer, especialmente em baixas energias de raios-x.
Emissão característica – A maioria dos elétrons que colidem com um alvo interagem com os elétrons orbitais do elemento alvo em interações não específicas e resultam em pouca ou nenhuma perturbação dos elétrons orbitais internos. No entanto, algumas interações resultam na ejeção de elétrons desses orbitais. As lacunas resultantes, ou buracos, representam estados instáveis de alta energia. Se as vacâncias orbitais estão nas camadas mais internas, os elétrons das camadas externas entram em cascata para preenchê-las e isso resulta em uma energia mais baixa e um estado mais estável. A energia liberada pelo processo pode se manifestar como raios-x. Cada uma das transições que podem ocorrer leva à emissão de linhas de raios X nítidas características do elemento alvo e da transição envolvida. Essas linhas de radiação características são emitidas com o contínuo.
Absorção de raios-X
Os raios X que incidem sobre uma amostra sofrem duas interações importantes com os elementos da amostra:absorção e dispersão. A absorção da radiação pode ocorrer por interações específicas que são consideráveis na excitação da amostra em espectrometria de raios X ou por interações mais gerais que influenciam a intensidade de raios X emitidos da amostra. A dispersão dos raios X leva à intensidade de fundo nos espectros observados.
Absorção de massa – Quando um feixe de raios X passa por um material, os fótons (campos eletromagnéticos) podem interagir de forma não específica com os elétrons dos orbitais dos elementos alvo, reduzindo a intensidade do feixe de raios X. As interações podem levar à ejeção fotoelétrica de elétrons ou dispersão do feixe de raios X. Em ambos os casos, o resultado geral é frequentemente descrito em termos de uma diminuição exponencial da intensidade com o comprimento do caminho do material absorvente. O coeficiente de absorção de massa é característico de um determinado elemento em energias especificadas de radiação x. Seu valor varia com o comprimento de onda da radiação x e o número atômico do elemento alvo.
O efeito fotoelétrico é o mais importante dos processos que levam à absorção dos raios X à medida que passam pela matéria. O efeito fotoelétrico é a ejeção de elétrons dos orbitais dos elementos no alvo de raios-x. Este processo é frequentemente o principal contribuinte para a absorção de raios X e é o modo de excitação dos espectros de raios X emitidos por elementos em amostras. Principalmente como resultado do processo fotoelétrico, o coeficiente de absorção de massa diminui constantemente com o aumento da energia da radiação X incidente. A curva absorção versus energia para um determinado elemento tem descontinuidades acentuadas. Estes resultam de energias características nas quais o processo fotoelétrico é especialmente eficiente.
Dispersão – Quando os fótons de raios X colidem com uma coleção de átomos, os fótons podem interagir com os elétrons dos elementos-alvo para resultar na dispersão dos fótons de raios X, conforme ilustrado na Fig 3. A dispersão dos raios X da amostra é a principal fonte de sinal de fundo nos espectros obtidos em espectrometria de raios-x. A dispersão dos raios X é causada principalmente por elétrons externos e fracamente retidos dos elementos. Se as colisões são elásticas, o espalhamento ocorre sem perda de energia e é conhecido como espalhamento Rayleigh. Se inelástico, o fóton de raios X perde energia para causar a ejeção de um elétron, e o espalhamento é incoerente. O caminho do fóton de raios X é desviado e o fóton tem uma perda de energia ou um comprimento de onda maior. Esta é a dispersão Compton.
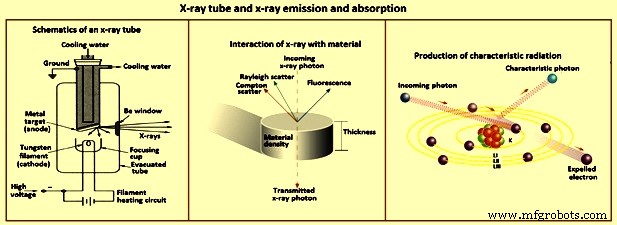
Fig 3 Tubo de raios X e emissão e absorção de raios X
A dispersão afeta a espectrometria de raios X de duas maneiras. Primeiro, a quantidade total de radiação espalhada aumenta com o número atômico devido ao maior número de elétrons. No entanto, amostras com matrizes de baixo número atômico mostram uma maior dispersão observada devido à redução da auto-absorção pela amostra. Em segundo lugar, a proporção da intensidade de dispersão 'Compton-to-Rayleigh' aumenta à medida que o número atômico da matriz da amostra diminui. A perda de energia associada ao espalhamento Compton resulta em uma mudança previsível no comprimento de onda da radiação.
Relações entre elementos e raios-x
As diferentes relações entre elementos e raios-x são mostradas na Fig 4.
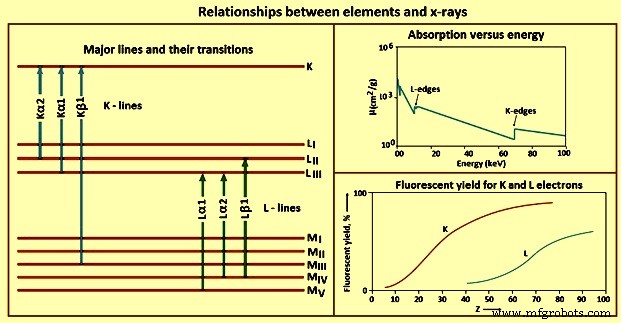
Fig 4 Relações entre elementos e raios-x
Absorção – Os fótons de raios X podem interagir com elétrons orbitais de elementos a serem absorvidos ou espalhados. A relação entre a absorção e o número atômico do elemento é importante na seleção das condições ideais de operação para a espectrometria de raios X.
Os coeficientes de absorção de massa diferem para um determinado elemento ou substância para cada elemento ou substância em uma determinada energia de raios X e em cada energia de raios X. Devido à maior probabilidade de interação com elétrons orbitais, o coeficiente de absorção de massa aumenta com o número atômico do elemento do material alvo. Em um determinado número atômico, o coeficiente de absorção de massa diminui com o comprimento de onda da radiação de raios X. Estas resultam de energias específicas necessárias para a ejeção fotoelétrica de elétrons dos diferentes orbitais do átomo e são características do elemento.
Bordas de absorção são descontinuidades ou pontos críticos no gráfico de absorção de massa versus comprimento de onda ou energia da radiação X incidente. A energia de borda de absorção é a quantidade exata que fotoejeta um elétron de um orbital de um elemento. Quanto menor o número quântico principal, maior é a energia necessária para ejetar um elétron dessa camada. O comprimento de onda de um raio-x que pode ejetar um elétron L é maior (de menos energia) do que o necessário para ejetar um elétron da camada K. Ou seja, a energia de borda de absorção K é maior que a energia de borda de absorção L para um determinado elemento.
Emissão – O efeito fotoelétrico é um mecanismo de absorção de raios X pelo qual são criados estados instáveis nos orbitais de elétrons dos átomos. Uma vez que as vacâncias nos orbitais internos são formadas, o relaxamento para o estado fundamental estável pode ocorrer pela emissão de raios X característicos do elemento excitado. A energia dos 1s O elétron é protegido do estado dos elétrons de valência, de modo que a energia de borda de absorção e a energia dos raios X emitidos são essencialmente independentes do estado de oxidação e da ligação do átomo.
K linhas – Uma vez que o efeito fotoelétrico cria uma vacância na camada K, o estado excitado relaxa preenchendo a vacância com um elétron de um orbital externo. Apenas certas transições são permitidas por causa de regras da mecânica quântica chamadas regras de seleção. As transições que seguem as regras de seleção são chamadas de linhas permitidas (diagrama), as que não seguem são chamadas de linhas proibidas, e aquelas que resultam em átomos com duas ou mais vacâncias nos orbitais internos no momento da emissão são chamadas de linhas satélite (não diagrama ) linhas. O número de K linhas, e a exata observada para um elemento, depende em parte do número de orbitais preenchidos.
Linhas L – Como a faixa de energia prática para a maioria dos espectrômetros de raios X WDXRF é de 0 keV a 100 keV e 0 keV a 40 keV para espectrômetros EDXRF, o uso de linhas de emissão diferentes das linhas K deve ser considerado. Para um dado elemento, as linhas L são excitadas com energia de raios X menor do que as linhas K. O uso de linhas L é particularmente valioso para elementos com números atômicos superiores a cerca de 45.
M linhas As linhas –M encontram aplicação limitada na espectrometria de raios X de rotina. As linhas não são observadas para elementos com números atômicos abaixo de 57 e, quando observadas, as energias de transição são baixas. O único uso prático para essas linhas é para elementos como tório, protactínio e urânio. Eles devem ser usados apenas nestes casos para evitar interferências com linhas L de outros elementos da amostra.
Rendimento fluorescente – Um elétron é ejetado de um orbital atômico pelo processo fotoelétrico com dois resultados possíveis:emissão de fótons de raios X ou ejeção de elétrons secundária (Auger). Um desses eventos ocorre para cada átomo excitado, mas não para ambos. Assim, a produção de elétrons secundários compete com a emissão de fótons de raios X de átomos excitados em uma amostra. A fração dos átomos excitados que emitem raios X é chamada de rendimento fluorescente. Este valor é uma propriedade do elemento e da linha de raios X em consideração. Elementos de baixo número atômico também têm baixo rendimento fluorescente. Juntamente com os altos coeficientes de absorção de massa que os raios X de baixa energia mostram, a detecção e determinação de elementos de baixo número atômico por espectrometria de raios X é um desafio.
Efeitos entre elementos – Para transições em espectrometria de raios X, nenhuma linha de emissão para uma determinada série (K, L, M) de um elemento tem energia igual ou maior que a borda de absorção dessa série. Um resultado importante é que os raios X emitidos por um elemento não podem fotoejetar elétrons do mesmo orbital de outros átomos daquele elemento. No entanto, uma amostra composta por uma mistura de elementos pode apresentar interações que são frequentemente chamadas de efeitos interelementos. Tais interações de elementos dentro de uma amostra frequentemente precisam de análise de dados especial.
Espectrômetros WDXRF
A instrumentação espectrométrica de raios-X introduzida comercialmente na década de 1950 é conhecida como dispersiva de comprimento de onda, que denota que a radiação emitida da amostra é colimada usando um colimador Soller e, em seguida, colide com um cristal analisador. O cristal difrata a radiação em diferentes extensões de acordo com a lei de Bragg e dependendo do comprimento de onda ou energia da radiação x. Esta dispersão angular da radiação permite a detecção sequencial ou simultânea dos raios X emitidos pelos elementos da amostra.
Instrumentos simultâneos normalmente contêm vários conjuntos de cristais e detectores de análise; um é ajustado para cada analito desejado na amostra. Apesar de caros, esses instrumentos são eficientes para a determinação rotineira de elementos pré-selecionados, mas não são facilmente convertidos para determinar outros elementos além dos selecionados na instalação.
Mais comuns são os instrumentos sequenciais que contêm um sistema mecânico conhecido como goniômetro que varia o ângulo entre a amostra, analisando o cristal e o detector. Desta forma, o comprimento de onda desejado de radiação x pode ser selecionado pelo movimento do goniômetro. Os espectrômetros sequenciais WDXRF podem ser controlados por computador para determinação automática de muitos elementos. As aplicações quantitativas de espectrômetros WDXRF automatizados são eficientes, pois o instrumento pode ser programado para ir para os ângulos corretos para as determinações desejadas. No entanto, as aplicações qualitativas são menos eficientes, pois o espectro deve ser varrido lentamente.
Tubos de raios-X – Várias fontes de energia podem ser usadas para criar os estados eletrônicos excitados nos átomos dos elementos que produzem a emissão de raios X. Entre estes estão os feixes de elétrons, feixes de partículas carregadas e radiação x. Feixes de elétrons são direcionados para a amostra em técnicas como microscopia eletrônica de varredura (MEV) e análise por microssonda eletrônica. No entanto, o uso de um feixe de elétrons precisa de um alto vácuo para evitar perdas de energia do elétron. A espectrometria de raios X é melhor usada como uma ferramenta analítica versátil do que como uma ferramenta especializada. Muitas amostras não são adequadas para um alto vácuo ou não são condutoras, o que causa problemas de carga elétrica quando sob um feixe de elétrons. Portanto, esta fonte de energia não é prática para espectrometria de raios X.
Isótopos radioativos que emitem radiação X são outra possibilidade de excitação de átomos para emitir raios X. No entanto, o fluxo de raios X de fontes isotópicas que podem ser manuseadas com segurança em um laboratório é muito fraco para uso prático. Como essas fontes normalmente emitem apenas algumas linhas estreitas de raios X, várias são necessárias para excitar muitos elementos com eficiência. A fonte de energia mais prática para espectrometria de raios X é um tubo de raios X (Fig. 3).
O espectrômetro WDXRF precisa de excitação eficiente de alta potência para ter um bom desempenho. Portanto, a estabilidade e a confiabilidade do tubo de raios X são importantes. Todos os componentes estão em alto vácuo. Um filamento é aquecido por uma tensão de filamento de 6 V a 14 V. O filamento aquecido emite elétrons termicamente. O fluxo de elétrons que flui entre o filamento e o ânodo alvo deve ser altamente regulado e controlado. Este fluxo de elétrons é corrente elétrica e é normalmente medido em mili-ampères. A corrente do tubo é muitas vezes referida como mA.
Um potencial de vários quilovolts é aplicado entre o filamento (cátodo) e o ânodo alvo, que serve como potencial de aceleração para os elétrons. Esta tensão é normalmente medida em quilovolts. O ânodo é normalmente de cobre e a superfície do alvo é revestida com depósitos de alta pureza de elementos como ródio, prata, cromo, molibdênio ou tungstênio. Os tubos de raios X usados para espectrometria WDXRF operam de 2 kW a 3 kW. Grande parte dessa energia se dissipa como calor, e são necessárias provisões para resfriamento de água do tubo de raios X. As fontes de alimentação e a eletrônica associada para esses tubos de raios X são grandes. Os elétrons atingem o alvo com uma energia cinética máxima equivalente ao potencial do tubo aplicado. Se a energia cinética do elétron exceder a energia de borda de absorção correspondente à ejeção de um elétron do orbital interno dos átomos do material alvo, o tubo emite linhas de raios X características do elemento alvo. A interação dos elétrons no feixe com os elétrons do elemento alvo também leva à emissão de um continuum. A área do contínuo e o comprimento de onda de intensidade máxima dependem do potencial, da corrente e da composição do ânodo.
Analisando cristais – Os raios X emitidos pelo tubo de raios X são direcionados para a amostra. Na maioria dos espectrômetros de raios X, a amostra é colocada acima do tubo de raios X no que é conhecido como óptica invertida. Isso facilita o posicionamento da superfície de um líquido usando a superfície inferior em vez da superior. A radiação X emitida pela amostra é colimada e colide com a superfície de um cristal analisador, que dispersa a radiação. O feixe paralelo de radiação X policromática da amostra é difratado de diferentes planos de rede no cristal. O reforço ocorre se a distância adicional que a radiação deve percorrer por difração de diferentes planos de rede é igual a um múltiplo inteiro do comprimento de onda. Se este não for o caso, ocorre interferência destrutiva. A lei de Bragg permite o cálculo do ângulo no qual um comprimento de onda deve ser selecionado para o cristal analisado.
Detetores – Detectores e eletrônicos associados no espectrômetro WDXRF detectam raios-x difratados do cristal analisador e rejeitam sinais indesejados, como difração de ordem superior ou inferior pelo cristal analisador ou ruído do detector. Dois detectores são normalmente posicionados em conjunto. O primeiro é um detector proporcional cheio de gás ou de fluxo de gás. Esses detectores consistem em um fio isolado da carcaça. Janelas finas de polímero na frente e atrás da caixa permitem a entrada e possível saída de radiação X. Um potencial de polarização de algumas centenas de volts é aplicado entre o fio e a carcaça.
Embora muitos gases possam ser usados, o gás típico é o P-10, uma mistura de 90% de argônio (Ar) e 10% de metano. Quando os raios X entram no detector, o argônio é ionizado para produzir muitos pares Ar+-e-. O fio anódico coleta os elétrons e os elétrons nas paredes catódicas do invólucro neutralizam os íons Ar+. O resultado é um pulso de corrente para cada fóton de raios X que entra no detector. Os detectores proporcionais preenchidos com P-10 são mais eficientes para detectar fótons de raios X de energias inferiores a cerca de 8 keV (comprimentos de onda superiores a cerca de 0,15 nm). A radiação X mais energética tende a passar pelo detector proporcional.
Um segundo detector frequentemente localizado atrás do contador proporcional é normalmente um detector de cintilação. Este detector consiste em um cristal de iodeto de sódio dopado com tálio [NaI(Tl)], que emite uma explosão de luz azul (410 nm) quando atingido por um fóton de raios X. O cristal é montado em um tubo fotomultiplicador que detecta os pulsos de luz. O número de fótons de luz produzidos é proporcional à energia do fóton de raios X incidente. Após o processamento eletrônico, a explosão de cintilação é convertida em um pulso de tensão proporcional em amplitude à energia do fóton de raios X. Esses dois detectores podem ser operados independentemente ou simultaneamente. Em operação simultânea, o potencial de operação do detector e o ganho de saída devem ser ajustados de modo que um fóton de raios X de uma determinada energia produza a mesma tensão de altura de pulso de ambos os detectores. Ambos os tipos de detectores precisam de cerca de 1 microssegundo para se recuperar entre os pulsos. Algumas contagens podem ser perdidas em taxas de fótons incidentes superiores a cerca de 30.000/s. A discriminação de altura de pulso dos pulsos de raios X do(s) detector(es) rejeita raios X de ordem superior ou inferior difratados do cristal analisador.
Fundamentos de operação – Quando uma amostra é considerada e o elemento analito selecionado, a primeira decisão é selecionar a linha de emissão. Na ausência de interferências específicas, normalmente é usada a linha mais energética plausível. Para elementos com números atômicos inferiores a cerca de 75, esta normalmente é a linha K, já que muitos espectrômetros WDXRF podem operar com potenciais de 100 kV para os tubos de raios-x. Quando possível, é selecionado um tubo de raios X que emite linhas características em energias logo acima da borda de absorção para a linha a ser usada para o elemento analito. Quando tal tubo não está disponível, a excitação deve ser realizada pelo uso do continuum para um tubo de raios X disponível.
O potencial do tubo de raios X deve ser definido em torno de 1,5 vezes a energia de borda de absorção ou superior. O detector deve ser selecionado com base na região de comprimento de onda a ser usada. O contador proporcional deve ser usado para raios X maiores que cerca de 0,6 nm, o detector de cintilação para comprimentos de onda menores que cerca de 0,2 nm e ambos para a região de sobreposição de 0,2 nm a 0,6 nm. Deve ser seleccionado um cristal de análise que permita detectar o comprimento de onda desejado. A maioria das seleções de parâmetros está sendo realizada por meio de controle por computador.
Espectrômetros de raios X com dispersão de energia
O uso de um goniômetro em espectrômetros de raios X WDXRF é baseado na necessidade de decompor em componentes os raios X emitidos por vários elementos em uma amostra. O uso de um dispositivo de dispersão é comum em muitos tipos de espectroscopia para realizar esta tarefa. Instrumentos sem os componentes mecânicos são desejáveis se a resolução adequada puder ser alcançada. O desenvolvimento de detectores de silício à deriva de lítio e sua aplicação à detecção de raios X em meados da década de 1960 levou a um campo de análise espectroscópica que ficou conhecido como espectrometria EDXRF.
Os tubos de raios X usados nos espectrômetros WDXRF são classificados em 2 kW a 3 kW e devem ser resfriados a água. Aqueles usados em espectrômetros EDXRF operam com potência muito menor e geralmente são resfriados a ar. Os tubos típicos variam de 9 W a 100 W. Diferentes materiais de anodo estão disponíveis, e cada fabricante de espectrômetros de raios X oferece recursos especiais de tubo de raios X. No entanto, após muitos testes de design de tubo, a maioria permanece com o design tradicional de 'janela lateral', embora seja muito menor do que os usados nos espectrômetros WDXRF. Um fator importante no projeto do tubo e fonte de alimentação associada é a estabilidade do tubo e da tensão.
Uma alternativa à excitação direta do tubo de raios X é o uso da excitação de alvo secundário. Neste modo, um tubo de raios X é usado para irradiar um alvo secundário, cuja fluorescência de raios X característica é usada por sua vez para excitar a emissão de raios X da amostra. Devido à perda substancial de eficiência ao usar um alvo secundário, são necessários tubos de raios X de potência mais alta do que o necessário para excitação direta.
A excitação do alvo secundário às vezes oferece vantagens significativas. Por exemplo, para determinar os baixos níveis de concentração de vanádio e cromo em uma amostra de ferro, esses elementos podem ser excitados com um alvo secundário de ferro sem excitação do ferro na amostra. Com excitação de tubo direto isso é difícil. Vários alvos secundários são necessários para cobrir uma ampla gama de elementos. O uso de excitação de alvo secundário tem sido apoiado como fonte de radiação monocromática para excitação. O significado dessa vantagem é que muitos dos programas de computador de parâmetros fundamentais, usados para calcular intensidades diretamente das equações básicas de raios X, precisam de radiação de excitação monocromática.
Na prática, a excitação do alvo secundário apenas se aproxima da radiação monocromática ideal. A excitação de tubo direto com filtros primários apropriados funciona bem quando comparada às técnicas de alvo secundário. Assim, a excitação direta do tubo de raios X continua sendo a mais prática para o maior número de aplicações de espectrometria dispersiva de energia (EDS). A principal força da técnica EDS está em suas capacidades de análise simultânea de múltiplos elementos. Embora ocorram casos especiais em que a excitação seletiva é desejável, isso frequentemente pode ser realizado com o uso inteligente de um tubo de raios X e filtro apropriados. Any fundamental design features which limit the simultaneous multi-element capability diminish the advantage of the EDXRF spectrometer.
Since direct x-ray tube excitation is the most common method used in EDS, there are factors which govern the selection of an x-ray tube. In wavelength-dispersive techniques, several x-ray tubes are normally available for the spectrometer. These can be changed for different applications. This is not normally the case with EDS-systems, since many WDXRF spectrometer has few if any choices of primary filters. In wavelength-dispersive techniques, it is customary to attempt to excite the desired element by the characteristic emission lines of the tube anode material, but the continuum is used more efficiently in EDXRF spectrometers. The use of EDXRF spectrometers has been enhanced by computer control of tube current and voltage and selection of the primary filter. Selection and efficient use of a single x-ray tube is important in the configuration of an EDXRF system.
Characteristic lines emitted by an x-ray tube have much larger intensity at their maxima than the continuous radiation emitted. These lines are to be used for excitation whenever possible. In addition, use of a primary filter between the x-ray tube and the sample can effectively approximate monochromatic radiation impinging on the sample from these characteristic lines. EDXRF spectrometers normally offer various x-ray tube anode materials. For selecting the x-ray tube anode material, the applications most likely to be encountered are to be considered.
The principal concern is to select an anode which has characteristic lines close to, but always higher, in energy than the absorption-edge energies to be encountered. None of the characteristic lines are to create spectral interference with elements to be determined. This includes consideration of such details as the Compton scatter peak for the characteristic lines. In addition, it is difficult to perform determinations of the element of the anode material. This is especially true with samples having low concentrations of that element.
Rhodium is a favourable tube anode material for general-purpose use. The characteristic lines of this element are efficient for the excitation of elements with absorption edges to around 15 keV. The excitation efficiency for the K lines of the transition elements is low. However, the continuum can be used efficiently in this region. Rhodium also has characteristic L lines at around 2.7 keV to 3.0 keV. These are efficient for the excitation of the K lines of low atomic number elements, such as aluminum, silicon, phosphorus, and sulphur. However, in these cases, a silver anode is preferable because of the Compton scatter radiation from the rhodium lines. The characteristic lines and the continuum from the x-ray tube can be used for excitation.
Although the elements of many samples can be excited effectively using a combination of the characteristic x-ray lines from the tube anode element and the continuum, more monochromatic radiation is sometimes desired. One such situation involves enhancing the use of fundamental-parameter computations which permit quantitative determination of elements without the need for several concentration standards. A more frequent situation is the need to reduce the background in the spectrum energy range to be used in the analysis. Use of primary filters placed between the x-ray tube and the sample can be effective in these cases and are normally incorporated under computer control in commercial spectrometers.
The object is to filter the primary radiation from the x-ray tube and selectively pass the characteristic lines of the anode element. This is accomplished using a filter made of the same element as the tube anode. Since x-rays of a given line (K, L, and so on) of an element are lower in energy than the absorption edge for that element, the photoelectric component of the mass absorption coefficient is small. Such a filter does not efficiently absorb the characteristic line emitted by the x-ray tube. The higher energy x-rays from the continuum are efficient for the photoelectric process in the filter and are highly attenuated by absorption. X-rays of lower energy than the filter material absorption edge are absorbed more efficiently as the energy decreases.
The result is x-radiation striking the sample with an intensity which is largely determined by the characteristic lines of the tube anode and that approximates monochromatic radiation. Increasing the thickness of the filter decreases the total intensity, with further gain in the monochromatic approximation.
Detectors – The selective determination of elements in a mixture using x-ray spectrometry depends upon resolving into separate components the spectral lines emitted by the different elements. This process needs an energy-sorting or wavelength-dispersing device. For the WDXRF spectrometer, this is accomplished by the analyzing crystal, which needs mechanical movement to select each desired wavelength according to Bragg’s law. Optionally, several fixed-crystal channels can be used for simultaneous measurement. In contrast, EDS is based on the ability of the detector to create signals proportional to the x-ray photon energy. Hence, mechanical devices, such as analyzing crystals, are not needed.
Several types of detectors have been used, including silicon, germanium, and mercuric iodide. The solid-state, lithium-drifted silicon detector [Si(Li)] was developed and applied to x-ray detection in the 1960s. By the early 1970s, this detector was firmly established in the field of x-ray spectrometry and was applied as an x-ray detection system for SEM and x-ray spectrometry. The Si(Li) detector provides excellent resolution. It can be considered as a layered structure. Under reversed bias of around 600 V, the active region acts as an insulator with an electric-field gradient throughout its volume.
When an x-ray photon enters the active region of the detector, photo ionization occurs with an electron-hole pair created for each 3.8 eV of photon energy. Ideally, the detector is to completely collect the charge created by each photon entry and result in a response for only that energy. Some background counts appear because of energy loss in the detector. Although these are kept to a minimum by engineering, incomplete charge collection in the detector contributes to background counts. From 1 keV to 20 keV, an important region in x-ray spectrometry, silicon detectors are efficient for conversion of x-ray photon energy into charge.
Analyzer systems – The x-ray spectrum of the sample is obtained by processing the energy distribution of x-ray photons which enter the detector. One x-ray photon entering the detector causes photo-ionization and produces a charge proportional to the photon energy. Several electrical sequences are to take place before this charge can be converted to a data point in the spectrum. A detailed knowledge of the electronics is not necessary, although an understanding of their functions is important. Upon entering the Si(Li) detector, an x-ray photon is converted into an electrical charge which is coupled to a field effect transistor (FET). The FET and the electronics comprising the preamplifier produce an output proportional to the energy of the x-ray photon. Using a pulsed optical preamplifier, this output is in the form of a step signal. Since photons vary in energy and number per unit time, the output signal, due to successive photons being emitted by a multi-element sample, resembles a staircase with various step heights and time spacing. When the output reaches a determined level, the detector and the FET circuitry reset to their starting level, and the process is repeated.
The preamplifier output is coupled to a pulse processor which amplifies and shapes the signal into a form acceptable for conversion to a digital form by an analog-to-digital converter (ADC). Amplification is necessary to match the analog signal to the full-scale range of the ADC. This process involves the energy calibration of the spectrometer. Drift in the gain and/or offset (zero) of the amplification results in errors in the energy assigned to the x-ray photons producing the signal. Hence, these calibrations are to be as stable as possible, and calibration is to be routinely checked.
The energy calibration is important for qualitative identification of the elements and for precise quantitative results when using spectrum-fitting programs. The amplifier provides gain and zero controls for calibrations. Normal operation in x-ray spectrometry is to set the time on the system clock to be used to acquire the spectrum. The processing of the pulses is not instantaneous. At high count rates, the time needed can become significant. When a pulse is detected and processing initiated, the clock is ‘stopped’ until the system is ready to process a new photon. The length of time the clock is off is called dead time; the time the clock is on is called live time. Their total is real time. The system monitors live time. If the spectrometer is operated with a 50 % dead time, the real time is twice the live time.
Processing of the pulse created by a photon is to be complete before another pulse occurs. A pulse pile-up rejector circuit blocks a pulse if it is received too soon. Once activated, the pulse pile-up rejector prevents the new signal from being processed if a second x-ray enters the detector before a prior pulse is fully processed. If analysis of the prior pulse had not yet been complete, it is also to be blocked from further processing. If this blockage is not performed, pulse pile-up occurs, resulting in an artifact which appears at energies equal to the sum of the photon energy of the first and second photons to enter the detector. These are frequently called sum peaks.
Despite pulse pile-up rejection circuitry, sum peaks are observed for intense peaks in the spectrum. This is the result of two photons entering the detector simultaneously or within a time difference faster than the fast discriminator can act. Sum peaks can be observed at twice the energy of an intense peak and / or at the sum of the energies of two intense peaks in the spectrum. Sum peaks decrease rapidly in intensity with count rate. The importance of electronic pulse-processing components to system performance is easily overlooked in EDS. However, stability, linearity, and proper calibration of these components are important for the use of the spectrometer.
EDXRF spectrometers require a dedicated computer system for data acquisition. Early spectrometers were heavy, unwieldy units which used hard-wired multichannel analyzers which could acquire data, but could do little to process it. Current spectrometer and data systems based on microprocessor technology are available as table-top units.
Fundamentals of operation – The simultaneous multi-element capability of EDS complicates the selection of optimum conditions because of the factors to be considered for each element. The compromises in spectroscopy are to be made, but the initial selection of instrument operating conditions can follow a logical sequence of decisions.
Qualitative analysis needs similar procedures, normally with less stringent requirements. Once a sample is received for analysis and the elements to be determined by x-ray spectrometry are identified, the next decision is to ascertain which x-ray lines are to be used for the determinations. As a general rule, K lines are used upto a K absorption-edge energy a few keV below the characteristic line of the x-ray tube anode element. The continuum can be used for excitation if the voltage to the x-ray tube is set sufficiently high to place the continuum maximum at energy higher than the absorption edge and if a back-ground filter is used. In these cases, K absorption-edge energies can be used upto around 66 % of the maximum operating kV of the x-ray tube. However, the observed peaks lie on a continuum background and reduce the signal-to-noise ratio.
For a 50-kV x-ray tube, absorption edges as high as 30 keV can be used if the element is present in sufficient concentration. For a 30-kV rhodium or silver tube, one is restricted essentially to excitation by the characteristic tube lines. This is of no great concern unless there is a special interest in the elements between atomic numbers 41 and 50 (niobium to tin). Elements above atomic number 50 (40 for a 30-kV system) are normally to be determined using the L lines of their x-ray spectra.
To excite all L lines, the incident x-ray photon energy is to exceed the LI absorption edge. For practical use, the energy of the L lines is to be higher than around l keV. For the L line spectra, this needs atomic numbers higher than 30. At such low x-ray energies, absorption of the x-rays and low fluorescent yield in the L emission in this region needs high concentration of the element to be determined and excellent sample preparation. Overlap of the K lines of the low atomic number elements in this region also causes difficulty. For example, the K lines of phosphorus overlap with the L lines of zirconium and the M lines of iridium at around 2 keV. These problems are to be considered, but are to a large degree solved by careful use of processing software.
Once the x-ray spectral lines are selected for determination of the elements, the next step is to decide whether all analyte elements in the sample can be determined with one instrumental setting. Although the multi-element capability of EDS is useful, all elements in every sample cannot be determined with a single set of instrument parameters. Some applications need more than one condition, such as a mixture of low atomic number elements and transition elements. The transition elements are best determined by excitation using the K lines of rhodium or silver and the low atomic number elements with the L lines or a properly adjusted continuum using a background filter. Computer control of instrument parameters facilitates changing the conditions. Whether automatic or manual control is used, all samples are to be analyzed under one set of conditions, then analyzed again using the alternate set. This is preferred over changing conditions between samples.
X-ray tube operating voltage affects the efficiency of excitation of each element in the spectrum and the integrated x-ray photon flux from the tube. The tube current affects the flux only. Hence, once the operating kV has been set, the tube current typically is adjusted until the system is processing counts efficiently. System dead time is to be maintained below, but near, 50 %. The voltage and current settings for the x-ray tube have a sensitive effect on the rate of information acquisition and count distribution among the respective spectral peaks for a given type of sample.
Selection of primary tube filter thickness is important. If the filter is changed, the tube current, and sometimes the voltage, frequently needs resetting since the filter alters the intensity distribution of the x-rays striking the sample. When characteristic tube lines are used for excitation, the filter is normally made from the tube anode element. The intensity of the transmitted x-rays decrease exponentially with increasing filter thickness. It is common to have two or three primary filters made from the tube anode element in the filter holder. The selection is to reflect optimum count rate corresponding with reasonable current and voltage settings. Thicker filters attenuate lower energy radiation more effectively and reduce the excitation efficiency for the element with low absorption coefficients.
The remaining decision is the choice of atmosphere in the sample chamber. If x-rays below around 5 keV are to be implemented, use of a vacuum can be advantageous. Intensity can increase sufficiently to reduce significantly the counting time needed to achieve an adequate number of counts. If the concentration of elements yielding these x-rays is sufficiently high, the vacuum may not be needed. Because of the extra precautions needed in sample criteria and handling, a vacuum path is not to be used unless significant benefit is realized. Similar reasoning applies to the helium atmosphere.
These guidelines are useful for initial selection of operating conditions. The instrumental parameters are interactive, and a change in one parameter needs adjustment of another. For example, selection of a thicker primary filter or a decrease in the tube voltage needs an increase in the tube current.
Sample preparation
The care taken to determine the best method of sample preparation for a given material and careful adherence to that method frequently determine the quality of results obtained. Sample preparation is the single most important step in an analysis, yet it is frequently given the least attention. In most cases, the stability and overall reproducibility of x-ray instrumentation are the least significant factor affecting the precision of analytical measurements. Frequently, the precision of analytical results expected from x-ray spectrometric determinations is expressed in terms of the theoretical statistics of measurement of x-ray intensities.
When replicate samples are prepared and actual standard deviations measured, deviations are found to be larger than those predicted by counting statistics. If precision is poor, any one analytical result can also be poor, since it can differ substantially from the ‘true’ value. The variety of sample types which can be analyzed using x-ray spectrometry necessitates different sample preparation techniques.
Samples are frequently classified as infinitely thick or infinitely thin based on measurement of the attenuation of x-rays. Samples are considered to be infinitely thick if further increase in the thickness yields no increase in observed x-ray intensity. The critical value for infinite thickness depends on the energy of the emitted x-radiation and the mass absorption coefficient of the sample matrix for those x-rays. For pure iron, the critical thickness is around 40 m for iron x-rays.
Although infinitely thin samples afford several advantages, it is rarely feasible to prepare them from routine samples. Many samples fall between these two cases and need extreme care in preparation. In addition to preparation of the sample, precise positioning of the sample in the spectrometer is critical for quantitative determinations.
Solid samples – These are defined as single bulk materials, as opposed to powders, filings, or turnings. Solid samples can frequently be machined to the shape and dimensions of the sample holder. The processing is not to contaminate the sample surface to be used for analysis. In other cases, small parts and pieces are to be analyzed as received. The reproducible positioning of these samples in the spectrometer is critical. It is frequently useful to fashion a wax mould of the part which fits into the sample holder. Using the mould as a positioning aid, other identical samples can be reproducibly placed in the spectrometer.
Samples taken from unfinished bulk material frequently needs surface preparation prior to quantitative analysis. Surface finishing can be done using a polishing wheel, steel wool, or belt grinder, with subsequent polishing using increasingly fine abrasives. Surface roughness less than 100 micrometers is normally sufficient for x-ray energies above around 5 keV, but surface roughness of less than 20 micrometers to 40 micrometers is needed for energies down to around 2 keV. Several precautions are necessary. Alloys of soft metals can smear on the surface as the sample is polished, resulting in a surface coating of the soft metal which yields high x-ray intensities for that element and subsequently high analytical results.
Polishing grooves on the surface of the sample can seriously affect the measured intensity of low-energy x-rays. This can be examined by repetitive measurement of the intensity of a sample after 45 degrees or 90 degrees rotation. Use of a sample spinner reduces this effect. If a sample spinner is not available, the sample is to be placed in the spectrometer such that the incident x-radiation is parallel to the polishing direction.
Powders and briquettes – Powder samples can be received as powders or prepared from pulverized bulk material too inhomogeneous for direct analysis. Typical bulk samples pulverized before analysis are ores, and refractory materials. Powders can be analyzed using the spectrometer, pressed into pellets or briquettes, or fused with a flux, such as lithium tetra borate. The fused product can be reground and pressed or cast as a disk. For precise quantitative determinations, loose powders are rarely acceptable, especially when low-energy x-rays are used. Pressed briquettes are more reliable. However, experience indicates that the best compromise is reground and pressed fusion products. This technique eliminates several problems associated with particle-size effects.
Particle-size effects result from the absorption of the incident and emitted x-rays within an individual particle. If the mass absorption coefficient of the sample matrix is high for the x-radiation used, particles even a few microns in diameter can considerably affect attenuation of the radiation within each particle. If the sample consists of particles of various sizes, or the particle size varies between samples, the resulting x-ray intensities can be difficult to interpret. This problem is compounded by the tendency of a material composed of a mixture of particle sizes to segregate when packed.
Determination of elements using low-energy x-radiation can lead to errors from particle-size effects of as much as 50 %. If the needed speed of analysis prohibits use of fusion techniques, direct determination from packed powders can be considered. The sample is to be ground, if possible, to a particle size below the critical value. The grinding time needed frequently can be ascertained by measuring the intensity from a reference sample at increasing grinding times until no further increase is observed. The lowest energy x-ray to be used in analysis is to be selected for this test.
Briquettes or pressed powders yield better precision than packed powder samples and are relatively simple and economical to prepare. In several cases, only a hydraulic press and a suitable die are needed. In the simplest case, the die diameter is to be the same as the sample holder so that the pressed briquettes fit directly into the holder. The amount of pressure needed to press a briquette which yields maximum intensity depends on the sample matrix, the energy of the x-ray to be used, and the initial particle size of the sample. Hence, prior grinding of the sample to a particle size which is less than 100 micrometers is advisable.
A series of briquettes are to be prepared from a homogeneous powder using increasing pressure. The measured intensity of the x-ray lines to be used in the analysis is plotted versus the briquetting pressure. The measured intensity is to approach a fixed value, perhaps asymptotically. Pressures of 138 MPa to 276 MPa may be needed. For materials which do not cohere to form stable briquettes, a binding agent is needed. Acceptable binding agents include powdered cellulose, detergent powders, starch, stearic acid, boric acid, lithium carbonate, polyvinyl alcohol, and commercial binders.
Briquettes which are not mechanically stable can be improved by pressing them into backing of pre-pressed binder, such as boric acid, or by the use of a die which presses a cup from a binding agent. The sample powder can then be pressed into a briquette supported by the cup. Improved results are frequently achieved if around 0.1 mm to 0.5 mm is removed from the surface of the briquette prior to the measurement.
Fusion of materials – Fusion of materials with a flux can be performed for several reasons. Some refractory materials cannot be dissolved, ground into fine powders, or converted into a suitable homogeneous form for x-ray spectrometric analysis. Other samples can have compositions which lead to severe inter-element effects, and dilution in the flux reduces these. The fused product, cast into a glass button, provides a stable, homogeneous sample well suited for x-ray measurements. The disadvantages of fusion techniques are the time and material costs involved as well as the dilution of the elements which can result in a reduction in x-ray intensity. However, when other methods of sample preparation fail, fusion frequently provides the needed results.
Low-temperature fusions can be carried out using potassium pyro-sulphate. More common are the glass-forming fusions with lithium borate, lithium tetra-borate, or sodium tetra-borate. Flux-to-sample ratios range from 1:1 to 10:1. The lithium fluxes have lower mass absorption coefficients and hence less effect on the intensity of the low-energy x-rays. An immense variety of flux-additive recipes are reported for various sample types. Lithium carbonate can be added to render acidic samples more soluble in the flux. Lithium fluoride has the same effect on basic samples. Lithium carbonate also reduces the fusion temperature. Oxidants, such as sodium nitrate and potassium chlorate, can be added to sulphides and other mixtures to prevent loss of these elements.
Filters and ion-exchange resins – Various filters, ion-exchange resin beads, and ion-exchange resin-impregnated filter papers have become important sampling substrates for samples for x-ray spectrometric analysis. Filter materials can be composed of filter paper, membrane filters, glass fiber filters, and so on. Filters are used in a variety of applications. One widely used application is in the collection of aerosol samples from the atmosphere. Loadings of several milligrams of sample on the filter can correspond to sampling several hundred cubic meters of atmosphere. Such sampling can be performed in any environment. Many elements can be determined directly on these filters by x-ray spectrometric analysis. Particulate samples collected in this way present problems, stemming primarily from particle-size effects, which are reduced in part by the need to collect two particle-size regions using dichotomous samplers. With these units, particles are separated into those smaller and those larger than around 2 micrometers in diameter.
The smaller particles tend to represent man-made materials; the larger ones are of natural origin. The smaller particles show fewer particle-size effects, and an x-ray spectrometric determination of even low atomic number elements, such as sulphur, is possible. Glass fiber filters are frequently used for this purpose. Filters can also be used for non-aerosol atmospheric components, such as reactive gases. Filter materials can be impregnated with a reagent reactive to the gas which traps it chemically. Sampling is accomplished by conveying atmospheric gases through a treated filter under carefully controlled conditions. An example is a damp filter treated with ferric ion solution used to trap hydrogen sulphide. The excess iron can be rinsed from the filter, but the precipitated ferrous sulphide remains. The sulphur can be determined directly or indirectly by measuring the iron x-radiation. The key to determining atmospheric components is the development of suitable standards.
Filters can be used to determine solution components in ways parallel to those described for atmospheric components. Particulate materials can be filtered directly from solution. For example, particulate materials in environmental water samples are defined as that which is filtered using a 0.45 micrometer pore diameter membrane filter. Hence, filtration of particles from water can be accomplished using such filters, and direct x-ray spectrometric analysis performed. Application of filter sampling to dissolved elements in water is becoming more common. The principle is similar to the reactive reagent-impregnated filter application to atmospheric gases. In some cases, the filter can be impregnated with ion-exchange resins which trap ions as the solution passes through the filter.
Procedures using ion-exchange resin-impregnated filters are to be carefully checked, since several passes of the solution can be needed, and distribution of the ions across the paper thickness is seldom uniform. However, for solutions, a reaction can be performed prior to filtration. For example, many ions can be precipitated quantitatively from aqueous solution, even at parts per billion concentration levels. The precipitates can be collected using 0.45 micrometers pore diameter membrane filters, which are then mounted between two Mylar sheets retained by ring clips on a standard plastic sample cup. Simultaneous multi-element determinations are then performed using XRF spectrometer.
Detection limits on the filters of as low as a few tenths of a microgram are common. If 100 g of sample solution is used, this corresponds to the detection limits of a few parts per billion in the sample. Standards are easily prepared as aqueous solutions. ‘Standard reference materials’ (SRM) for environmental waters and industrial effluent water are available.
Thin-film samples – Thin-film samples are ideal for x-ray spectrometric analysis. The x-ray intensity of an infinitely thin sample is proportional to the mass of the element on the film, and the spectral intensities are free of inter-element and mass absorption coefficient effects. However, in practice, perfect thin-film samples are rarely encountered. Powder samples of sufficiently small and homogeneous particle size can be distributed on an adhesive surface, such as cellophane tape, or placed between two drum-tight layers of Mylar film mounted on a sample cup.
More important thin-film types are platings and coatings on various substrates. Analysis of these sample types is increasingly important for the electronics industry. Of particular concern are measurements of film thickness and composition. Several techniques can be used, including the substrate intensity attenuation method, the coating intensity method, various intensity ratio methods, and the variable takeoff angle method. The last method is not practical in most commercial spectrometers. To be infinitely thin to most x-rays used in x-ray spectrometric analyses, the samples are to be 10 micrometers to 200 micrometers thick.
Liquids – Liquids can also be analyzed using x-ray spectrometry. The design of x-ray spectrometric instrumentation using inverted optics, in which the sample is above the x-ray source and detector, facilitates the use of liquid samples. This convenient geometry demands caution in the preparation of liquid samples to avoid damaging the source or detector by such accidents as spills and leaking sample cups.
Quantitative standards are easily prepared for liquid samples. However, since solvents are normally composed of low atomic number elements, the Rayleigh and Compton scatter intensity is high, which increases background and leads to high limits of detection. These problems can be minimized by use of suitable primary tube filters, which reduce the scattered x-radiation in the analytically useful region.
Care is to be taken with liquids containing suspended solids. If the suspension settles during the measurement time, the x-ray intensity of the contents of the sediment is enhanced. The x-ray intensity from solution components or homogeneous suspension can decrease as a result of sediment absorption, which leads to erroneous results. This possibility is tested by brief, repetitive measurements, beginning immediately after a sample is prepared. Any observed increase or decrease in intensity with time indicates segregation in the sample. In these cases, an additive which stabilizes the suspension can be used, or the suspended content can be collected on a filter for analysis.
Special sample types – Applications of x-ray spectrometric analysis do not always provide convenient samples which can fit one of the above categories. Non-destructive analyses are occasionally needed on production products which are not 32 mm diameter circles of infinite thickness. Examples include computer disks, machined parts, and long, coated strips or wire. In these cases, a sample compartment which accommodates the samples can frequently be designed. With the development of the mercuric iodide detector, which can provide adequate resolution for many analyses without a liquid nitrogen dewar, special analytical systems for on-line and non-destructive analysis of large samples can become increasingly feasible.
Processo de manufatura
- Análise da forma de onda
- Opções de análise
- Exemplo de circuitos e listas de rede
- Análise de falha de componente
- Análise de falha de componente (continuação)
- O que é análise de rede?
- Mais sobre análise de espectro
- Óculos de raio-X
- Análise de potência orientada por software
- Inspeção Automatizada de Raios-X