


Modelagem e simulação propõe novos insights para SARS-CoV-2
Em nosso blog anterior, discutimos o uso de ferramentas de modelagem preditiva para construir estruturas iniciais de nível atômico de potenciais alvos de drogas (por exemplo, proteínas) e para refinar regiões que não eram experimentalmente determináveis ( Ver Vídeo ) Essas ferramentas incluem a adição de átomos de hidrogênio e loops flexíveis que às vezes não podem ser resolvidos experimentalmente. Nós examinamos isso no contexto da microscopia crioeletrônica (crio-EM) da proteína SARS-CoV-2 spike (S) publicada recentemente na revista Science (DOI:10.1126 / science.abb2507).
Neste blog, iremos detalhar como a modelagem molecular e simulação de modelos estruturais refinados, como a proteína SARS-CoV-2 S, podem auxiliar na geração de novas hipóteses para a descoberta e desenho de supostas terapêuticas para tratar COVID-19.
A ligação de drogas depende de mudanças estruturais
Em sistemas vivos, as proteínas existem naturalmente como entidades dinâmicas. Sua dinâmica freqüentemente predetermina sua função. O físico Richard Feynman disse uma vez:
“Se fôssemos nomear a suposição mais poderosa de todas, que nos leva continuamente a uma tentativa de entender a vida, é que todas as coisas são feitas de átomos e que tudo o que os seres vivos fazem pode ser entendido em termos dos balanços e balanços dos átomos. ” 1
A proteína SARS-CoV-2 S não é exceção ao princípio de Feynman. Antes de entrar nas células humanas, a proteína S se liga a um receptor conhecido como enzima conversora de angiotensina 2 (ACE2). 2 O domínio de ligação ao receptor (RBD) é parte da proteína S que se liga a ACE2 ( Figura 1A ) O RBD pode existir em pelo menos dois estados conformacionais primários chamados de estados para cima (acessível ao receptor) e para baixo (inacessível para o receptor) ( Figura 1B ) Quando o RBD está no estado up, a proteína S está mais “aberta” para facilitar a ligação de ACE2. 2 Estudos sugeriram que o estado inativo, inacessível ao receptor, é mais estável. 2 Isso implica que a terapêutica putativa, como pequenas moléculas orgânicas capazes de se ligar a RBD, poderia estabilizar o RBD no estado negativo e impedir que o vírus interagisse com ACE2; assim, impedindo o COVID-19 de infectar pessoas.
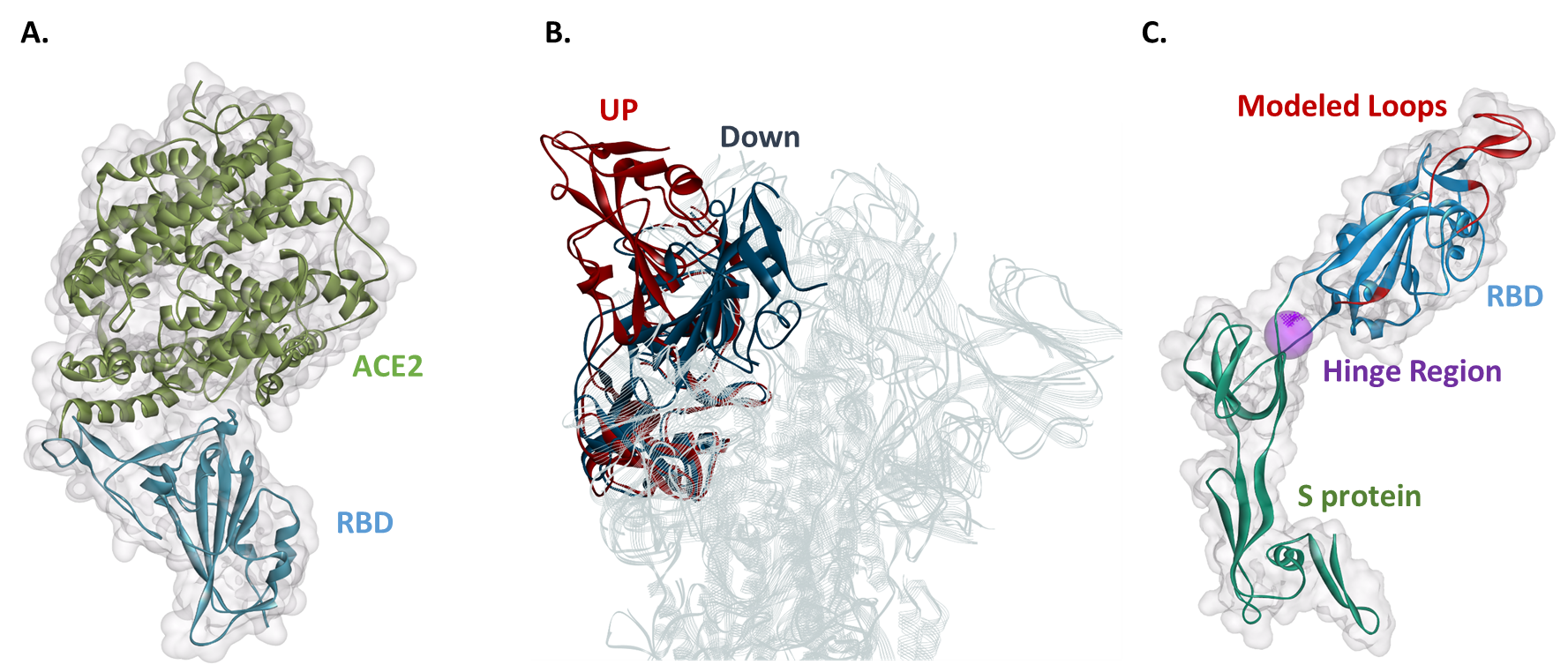
O RBD da proteína S é como a dobradiça de uma porta
Um ligante flexível conecta o RBD à proteína S restante. A flexibilidade do linker permite que o RBD faça a transição do estado de baixo para cima por meio de um movimento de dobradiça ( Figura 1B ) Da proteína SARS-CoV-2 S, extraímos o RBD com o ligante flexível associado e domínio adjacente ( Figura 1C ) Usamos a estrutura RBD da proteína S no estado up (PDB 6VYB), mas esta estrutura carece de três loops potencialmente importantes para a ligação de ACE2 ( Figura 1C ) Como resultado, tivemos que construir um modelo de homologia do RBD com o ligante flexível usando a estrutura crio-EM da qual o truncamento foi feito originalmente (PDB 6VYB) com um modelo adicional. O modelo adicional era a estrutura cristalina do RBD sozinho no complexo com ACE2 (PDB 6M17) ( Figura 1A )
A estrutura RBD contém loops incluindo um mais de 20 resíduos de aminoácidos, que não estão presentes na estrutura de estado aberto (PDB 6VYB). Dois desses loops formam a interação com ACE2; assim, a modelagem de homologia é necessária para entender as interações da proteína Spike. Podemos então atribuir átomos de hidrogênio em estados de protonação apropriados para simular condições fisiológicas como o pH.
Poderíamos então realizar uma simulação de dinâmica molecular (MD) para simular a transição conformacional e / ou prever possíveis locais de ligação onde supostas moléculas pequenas podem se ligar para interromper a interação da proteína S com ACE2. Quando previmos possíveis locais de ligação usando BIOVIA Discovery Studio para nosso modelo de homologia do RBD com o ligante flexível, identificamos um local de ligação localizado na dobradiça do ligador flexível ( Figura 1C ) Nós rotulamos essa região de dobradiça e observamos que pode valer a pena uma investigação mais aprofundada para a descoberta de possíveis terapêuticas. Se uma pequena molécula se ligasse à região de dobradiça, ela poderia travar o RBD em um estado para baixo e, assim, impedir a ligação de ACE2.
Outras investigações
Investigações extensivas, incluindo previsões computacionais e experimentos biológicos, podem esclarecer ainda mais a utilidade da região de dobradiça. Exemplos de previsões computacionais podem incluir análise de modo normal (NMA) e / ou simulação MD. 3 Por exemplo, uma longa escala de tempo, aproximadamente centenas de nanossegundos, de simulação MD poderia permitir aos cientistas amostrar múltiplas conformações da região de dobradiça.
Por outro lado, um NMA pode fornecer uma estimativa grosseira e rápida das transições conformacionais. 3 Conformações específicas da simulação MD e / ou NMA são pontos de partida para a triagem virtual de alto rendimento de bancos de dados de moléculas pequenas em potencial. Os cientistas poderiam encaixar e pontuar cada pequena molécula em todas as conformações. Eles poderiam então classificar todas as poses resultantes e enviar os melhores acertos para validação experimental. Estudos têm mostrado que este método de descoberta computacional de drogas, muitas vezes referido como triagem virtual baseada em conjunto, aumenta as chances de identificar candidatos a drogas. 4 O método também reflete a realidade de que a ligação à droga depende de mudanças estruturais na proteína, conforme observado acima. Acreditamos que os resultados preliminares identificados aqui são interessantes e dignos de uma investigação mais aprofundada.
Em segundo lugar, gostaríamos de observar que o modelo estrutural da proteína S refinado pode ser usado como um alvo para imunoterapêuticos. 5 Os cientistas podem criar anticorpos monoclonais que se ligam às proteínas SARS CoV-2 S com base no conhecimento prévio do sítio de ligação ACE2. Eles poderiam então executar in silico estudos de afinidade-maturação para melhorar a especificidade da ligação. 6
Como um apoiador ativo da comunidade científica que está colaborando hoje nas soluções COVID-19, a BIOVIA Dassault Systèmes desenvolve o BIOVIA Discovery Studio. Este ambiente comprovado de modelagem e simulação de ciências biológicas reúne mais de 30 anos de pesquisa revisada por pares e in silico de classe mundial técnicas. O software fornece aos cientistas um conjunto de ferramentas completo para uso desde a identificação de alvos até a otimização de chumbo, incluindo ferramentas para design e análise de produtos biológicos, simulações clássicas, design baseado em estrutura e fragmento, triagem de ligante virtual, bem como ADME e previsão de toxicidade.
Como parte da responsabilidade social corporativa da Dassault Systèmes, a BIOVIA tem o prazer de oferecer grupos de pesquisa acadêmica qualificados envolvidos em estudos relacionados ao SARS-CoV-2 uma licença gratuita de seis meses para o BIOVIA Discovery Studio para auxiliá-los na busca de candidatos a drogas terapêuticas rápidas, seguras e eficazes contra o vírus SARS-CoV-2. Se você é um pesquisador acadêmico nesta área, solicite uma licença de software e faça o download. Esta oferta será válida até 30 de junho de 2020.
biológico
- Pixus:novas placas frontais grossas e robustas para placas incorporadas
- GE apresenta novo produto para aplicativos de controle e monitoramento
- DSM e Nedcam desenvolvem novos aplicativos para impressão 3D de tamanho grande
- Teradyne planeja novo hub Cobot para empresas de portfólio UR e MiR
- PLASTICS lança novo padrão de segurança para robótica e moldagem por injeção
- Um Novo Roteiro para Cadeias de Abastecimento de Petróleo e Gás
- Coaching para Sustentabilidade:Implementando e Sustentando Novos Processos e Mudanças
- B&R apresenta nova ferramenta de simulação para desenvolvimento de gêmeos digitais
- ABB fornece planejamento de automação e eletrificação para nova mina na Suécia
- 5G e Edge levantam novos desafios de segurança cibernética para 2021