


Adsorção de metais de transição em fosforeno preto:um estudo de primeiros princípios
Resumo
O fosforeno preto é um novo material bidimensional que possui propriedades únicas e amplas aplicações. Usando cálculos de primeiros princípios, investigamos o comportamento de adsorção de 12 metais de transição diferentes (TMs; Fe, Co, Ni, Cu, Ru, Rh, Pd, Ag, Os, Ir, Pt e Au) em fosforeno. Nossos resultados mostraram que todos os sistemas de adsorção possuem uma grande energia de ligação. Os sistemas de Fe-, Co- e Au-fosforeno exibem estados magnéticos com momentos magnéticos de 2, 1 e 0,96 μ B , respectivamente, o que significa que esses sistemas são semicondutores magnéticos. A adsorção de moléculas de oxigênio em fosforeno-TM também foi investigada. Curiosamente, todos os O 2 - Sistemas (TM-fosforeno), exceto O 2 - (Pd-fosforeno), pode alongar a ligação O – O, que é crítica para sua aplicação como catalisadores na oxidação de CO. Também descobrimos que a adsorção de O 2 moléculas permitem o O 2 - (Fe-, Ni, Cu-, Ir-, Rh-, Ag- e Au-fosforeno) sistemas se tornem semicondutores magnéticos, e permite O 2 - (Co-fosforeno) para exibir o estado semimetálico. Espera-se que nossos resultados tenham implicações importantes para a catálise à base de fosforeno e spintrônica.
Histórico
Fosforeno [1,2,3], uma monocamada de átomos de fósforo dispostos em uma estrutura de favo de mel enrugada, tem propriedades únicas que incluem uma natureza semicondutora direta [4], mobilidade ultraelevada em temperatura ambiente [4,5,6], flexibilidade mecânica superior [7], e alto desempenho termoelétrico [8,9,10]. Essas propriedades tornam o fosforeno um material muito adequado para uma variedade de aplicações, como transistores de efeito de campo [1,11,12,13,14,15,16], baterias de íons de lítio e íon [17,18,19], células solares [20, 21], fotocatalisadores [22], spintrônica [23] e sensores de gás [24,25,26]. Porém, o fosforeno é um material não magnético, e algumas estratégias devem ser adotadas para ampliar sua aplicação.
Para materiais bidimensionais (2D), a adsorção é geralmente selecionada como a abordagem para induzir o magnetismo para aplicações específicas. Anteriormente, Cao et al. [27] mostraram que as propriedades eletrônicas e magnéticas do grafeno podem ser efetivamente moduladas por adátomos de Fe, Co, Ni e Cu. Kaloni et al. [28] demonstraram que momentos magnéticos podem ser induzidos em sistemas de siliceno decorados com Ti-, V-, Cr-, Mn-, Fe- e Co-decorados usando cálculos de primeiros princípios. Ersan et al. [29] descobriu que b -Arsenene exibia caracteres polarizados por spin após a adsorção de átomos de H, B, C, P, Ge, As e Sb. Além disso, w -Arseneno pode atingir momentos magnéticos líquidos com os adátomos de H, B, N, P, Cl, Ti, As e Sb. Para o fosforeno preto, Kulish et al. [30] previu que Ag-, Au-, Ti-, V-, Cr-, Mn-, Fe-, e Co-fosforeno são bastante estáveis, e uma gama diversificada de momentos magnéticos pode ser induzida em cálculos teóricos. Além disso, as propriedades de diferentes tipos de portadores de carga também podem ser ajustadas pela adsorção de diferentes átomos no fósforo. Ding e Wang [31] usaram os cálculos dos primeiros princípios para ilustrar sistematicamente as propriedades estruturais, eletrônicas e magnéticas dos átomos adsorvidos no fósforo. Eles notaram que os adátomos podem introduzir magnetismo no fosforeno, com os adátomos P, Co e Au induzindo propriedades magnéticas estáveis. Hu e Hong [32] usaram os cálculos dos primeiros princípios para demonstrar as propriedades magnéticas dos adátomos de metal no fosforeno; eles mostraram que o magnetismo pode ser obtido no fósforo pela adsorção de átomos de Cr, Fe, Co ou Au em sua superfície. Além disso, eles previram que o sistema de adsorção de Fe-fosforeno será um material semicondutor magnético diluído promissor. Assim, pode-se esperar que a adsorção de metais de transição (TMs) em fosforeno preto ajuste efetivamente as propriedades magnéticas do material.
Embora as investigações acima tenham estudado o comportamento de adsorção de metais de transição em fosforeno preto, alguns problemas permanecem sem solução. Por exemplo, estudos anteriores focaram principalmente nas propriedades de 3d TMs adsorvidos em fosforeno. Como as TMs 4d e 5d projetarão as propriedades do fosforeno? Além disso, metais nobres absorvidos em fosforeno também podem ser usados como catalisadores de átomo único. Li et al. [33] sugeriram que o siliceno com Au adsorvido pode ser um catalisador de alta atividade com baixas barreiras de energia catalítica para a oxidação de CO. Um metal nobre absorvido em fosforeno também pode ser um bom candidato para a oxidação de CO? Para responder a essas perguntas, apresentamos neste artigo os resultados de um estudo detalhado dos primeiros princípios sobre as propriedades estruturais, magnéticas e eletrônicas de 12 tipos diferentes de átomos de metal de transição adsorvidos em fosforeno preto. Selecionamos Fe, Co e Ni elementais, que são metais ferromagnéticos em sua fase principal; Cu elementar, que é diamagnético; e os metais nobres Ru, Rh, Pd, Ag, Os, Ir, Pt e Au, que são muito eficazes para a oxidação de CO [19, 34,35,36,37,38,39,40,41,42 , 43,44,45]. Descobrimos que o fosforeno forma ligações fortes com todos os 12 metais e que todos os sistemas de fosforeno-TM são bastante robustos. As propriedades eletrônicas e magnéticas do fosforeno podem ser efetivamente ajustadas pelos adátomos. Além disso, também descobrimos que a maioria dos sistemas de adsorção de fosforeno-TM são bons candidatos para o catalisador na oxidação de CO. Os resultados desta investigação podem ser usados para estudos fundamentais de fosforeno, e também podem ampliar sua aplicação potencial em muitos campos importantes .
Métodos / Experimental
Nossos cálculos foram baseados na teoria do funcional de densidade polarizada por spin (DFT), e foram realizados usando o Vienna Ab Initio Simulation Package (VASP) [46, 47] e a aproximação de gradiente generalizado (GGA) do Perdew-Burke-Ernzerhof ( PBE) funcional [48,49,50]. O método DFT-D3 de Grimme [51] foi usado para calcular a interação de van der Waals. Um corte de energia de 400 eV com um conjunto de base de onda plana foi empregado. Nos cálculos, os átomos foram relaxados até que a energia total convergisse para 1 × 10 −5 eV e a força residual em cada átomo era inferior a 0,01 eV / Å. Uma grande supercélula (4 × 3) ao longo das direções do zigue-zague e da poltrona foi usada para evitar interações entre as células unitárias vizinhas. As constantes de rede foram definidas como a =13,20 Å e b =13,74 Å. Aplicamos um espaço de vácuo de 20 Å no z direção para minimizar as interações entre camadas intermediárias adjacentes. Durante a otimização, um Monkhorst-Pack [52] k - grade de pontos de 3 × 3 × 1 foi adotada, e um k A grade de pontos de 7 × 7 × 1 foi usada para os cálculos de energia total.
Resultados e discussão
Exploramos primeiro as propriedades estruturais do fosforeno puro. A Figura 1a mostra as ilustrações das vistas superior e lateral da estrutura cristalina. Pode-se observar que a monocamada de fosforeno consiste em dois planos atômicos, e a célula unitária do fosforeno consiste em quatro átomos de P. A monocamada de fosforeno tem uma rede tetragonal com constantes de rede de equilíbrio a =3,30 Å e b =4,58 Å. O comprimento da ligação P – P na direção horizontal ( l 1 ) é 2,22 Å, enquanto o comprimento na outra direção ( l 2 ) é 2,26 Å. O fosforeno puro tem um bandgap direto de 0,89 eV (Fig. 1b), com a banda de condução mínima (CBM) e a banda de valência máxima (VBM) localizadas no ponto Г. A constante de rede e o bandgap que obtivemos estão de acordo com os valores obtidos em pesquisas anteriores [30,31,32,53].
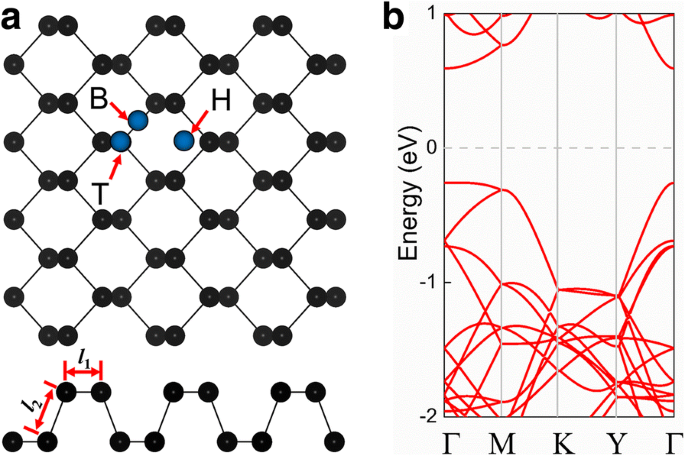
a Vistas superior e lateral da estrutura cristalina de fosforeno puro (4 × 3 × 1 supercélula). Os círculos azuis representam as posições típicas de um átomo de impureza adsorvido em um ponto oco (H), em uma ponte (B) entre dois átomos de fósforo e no topo de um átomo de fósforo (T). b Estrutura de banda eletrônica e primeira zona de Brillouin de fosforeno puro; o nível de Fermi é definido como zero
Um adatom típico é sempre adsorvido em qualquer uma das três posições:acima de um local oco (H), em uma ponte (B) entre dois átomos de fósforo e no topo de um átomo de fósforo (T). Calculamos a energia de adsorção de um adatom em fosforeno para examinar a estabilidade dos sistemas de adsorção usando a relação:
$$ {E} _ {\ mathrm {ad}} =\ left ({E} _ {\ mathrm {TM}} + {E} _ {\ mathrm {phosphorene}} \ right) - {E} _ {\ mathrm {TM} - \ mathrm {fósforo}} $$ (1)
onde E TM é a energia de um átomo de metal isolado, E fosforeno é a energia total da camada de fosforeno puro, e E TM-fosforeno é a energia total do sistema de adsorção. Com base nesta equação, uma energia de adsorção maior indica uma estrutura mais estável. Descobrimos que todos os átomos de metal estudados em nosso trabalho preferem ficar no local H do fosforeno. As energias de adsorção calculadas dos átomos de metal adsorvidos no sítio H do fosforeno, mostradas na Tabela 1, variam de 2 a 6 eV. O comprimento da ligação de fosforeno-TM ( d TM-P ) foi demonstrado ser curto, no intervalo de 2,11–2,43 Å. A análise de carga de Bader [54,55,56] mostra que 0,16, 0,16, 0,07, 0,17, 0,32, 0,33 e 0,16 | e | são transferidos dos átomos de metal Ru, Rh, Pd, Os, Ir, Pt e Au, respectivamente, para fosforeno nos sistemas de adsorção de (4d-TM) -fosforeno e (5d-TM) -fosforeno. Todos esses resultados denotam a formação de ligações químicas entre o adatom TM e o fosforeno. Além disso, esses resultados estão próximos de estudos recentes [30,31,32].
Conforme mostrado na Tabela 1, os sistemas Ni-, Cu-, Ru-, Rh-, Pd-, Ag-, Os-, Ir- e Pt-fosforeno exibem estados não magnéticos, enquanto o Fe-, Co- e Au -sistemas de fosforeno têm os momentos magnéticos de 2, 1 e 0,96 μ B , respectivamente. A densidade de carga polarizada por spin ( ρ = ρ spin-up - ρ redução da rotação ) também é mostrado na Fig. 2 para explorar a origem e distribuição do magnetismo nos sistemas de adsorção de fosforeno-TM magnético. O momento magnético em cada um desses casos origina-se principalmente do adatom, com um pequeno momento magnético resultante dos vizinhos mais próximos. Além disso, as estruturas de banda calculadas dos sistemas de Fe-, Co- e Au-fosforeno são representadas na Fig. 2. Pode-se ver que esses sistemas são todos semicondutores magnéticos com bandgaps de 0,38, 0,22 e 0,06 eV, respectivamente, que são úteis para aplicações spintrônicas.
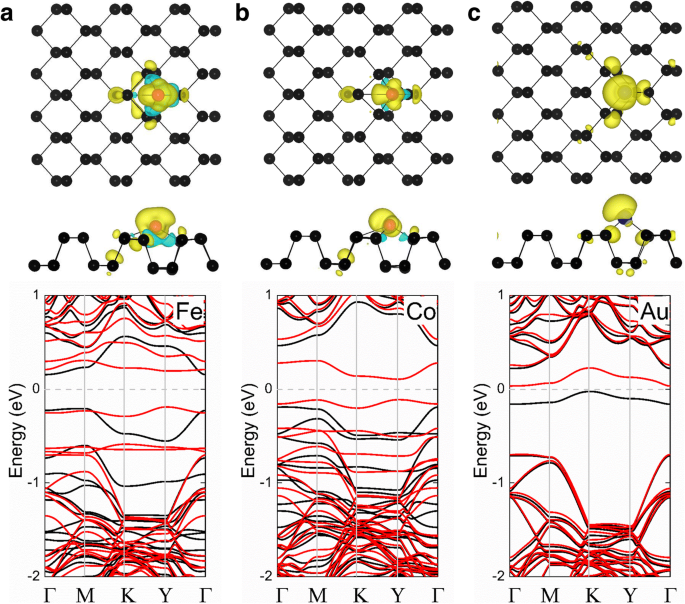
Densidades de spin do a Fe-fosforeno, b Co-fosforeno e c Os sistemas de Au-fosforeno são mostrados na linha superior; a estrutura de banda correspondente de cada sistema é mostrada na linha inferior. As esferas pretas e vermelhas representam átomos P e TM, respectivamente. Na linha superior, um gráfico da densidade de carga polarizada por spin com um valor de iso-superfície de densidade de carga de 0,002 e / Å 3 é sobreposta nas vistas superior e lateral da estrutura cristalina do fosforeno puro para cada um dos sistemas TM-fosforeno; as regiões amarela e ciano correspondem aos spins para cima e para baixo, respectivamente. No gráfico de estruturas de banda (linha inferior), as linhas pretas e vermelhas denotam canais de spin-up e spin-down, respectivamente; o nível de Fermi é definido como zero e é indicado pela linha tracejada cinza
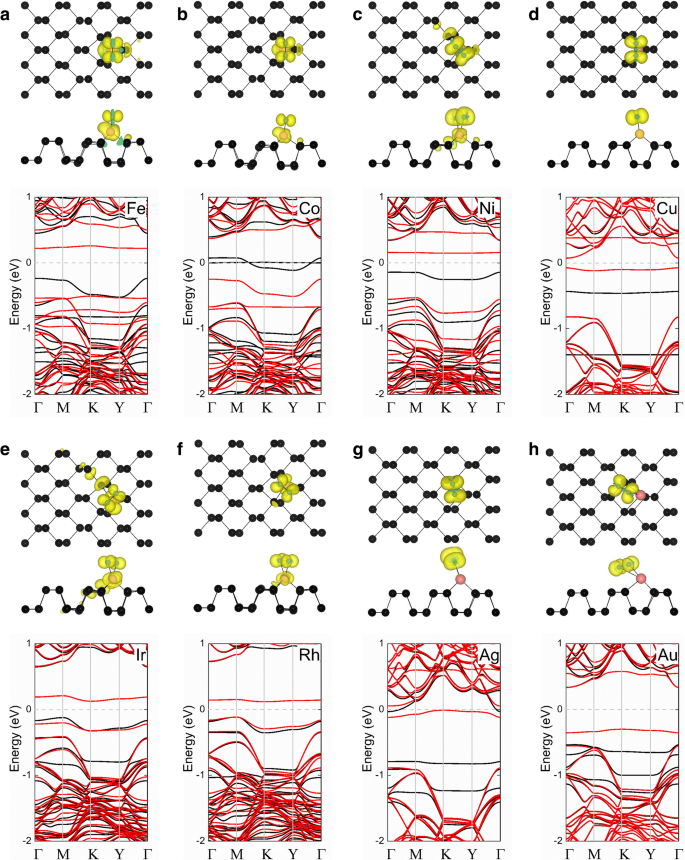
A seguir, estudamos o comportamento de adsorção de O 2 no topo do átomo TM nos sistemas fosforeno TM. Duas configurações típicas de menor energia para a adsorção de O 2 em sistemas TM-fosforeno (O 2 - (TM-fosforeno)) são mostrados na Fig. 3. Para O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Pd-fosforeno) e O 2 - (Pt-fosforeno) sistemas, o O 2 A molécula é paralela à direção do ziguezague do fosforeno (Fig. 3a), com um comprimento de ligação O – P de 1,84 Å, 1,86 Å, 2,04 Å, 2,18 Å e 2,05 Å, respectivamente. Para o O 2 - (Ni-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno) e O 2 - Sistemas (Au-fosforeno), a molécula está ao longo da direção em zigue-zague do fosforeno (Fig. 3b), a um certo ângulo da superfície. Enquanto isso, os dois átomos O vizinhos em torno do adatom TM não são equivalentes. Os resultados são exibidos na Tabela 2. A energia de adsorção ( E anúncio ) de O 2 em um O 2 - O sistema (TM-fosforeno) foi calculado como:
$$ {E} _ {\ mathrm {ad}} ={E} _ {\ mathrm {TM} - \ mathrm {fósforo}} + {E} _ {{\ mathrm {O}} ^ 2} - {E } _ {{\ mathrm {O}} ^ 2- \ mathrm {TM} - \ mathrm {fosforeno}} $$ (2)
onde \ ({E} _ {{\ mathrm {O}} ^ 2- \ mathrm {TM} - \ mathrm {fósforo}} \), E TM-fosforeno , e \ ({E} _ {{\ mathrm {O}} ^ 2} \) são as energias totais do O 2 - sistema (TM-fosforeno), o sistema TM-fosforeno e o O 2 molécula, respectivamente. Conforme mostrado na Tabela 2, as energias de adsorção são 2,659, 1,850, 0,970, 0,906, 2,402, 1,548, 0,001, 0,786, 3,109, 1,980, 0,416 e 1,029 eV para o O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Pd-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Pt-fosforeno) e O 2 - (Au-fosforeno) sistemas, respectivamente. Em todos os casos, as grandes energias de adsorção, exceto para o O 2 - (Pd-fosforeno) sistema indica que O 2 é quimicamente absorvido.
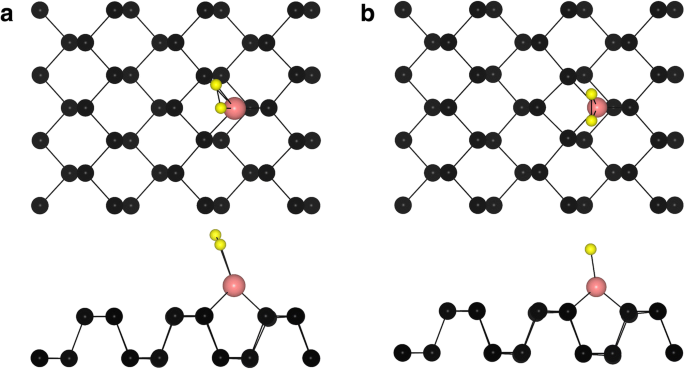
Vistas superior e lateral de sites de adsorção típicos de um O 2 molécula em TM-fosforeno. As esferas preta, rosa e amarela representam átomos P, TM e O, respectivamente
É bastante reconhecido que o alongamento da ligação O – O é crucial para os mecanismos Langmuir-Hinshelwood e Eley-Rideal de um catalisador na oxidação do CO [57]. De modo geral, quanto maior o comprimento da ligação O – O, mais fácil será a reação do catalisador. Os comprimentos de ligação O – O e TM – O em cada sistema também são mostrados na Tabela 2. Obviamente, a ligação O – O aumenta de 1,23 Å para o prístino O 2 molécula para 1,38, 1,36, 1,32, 1,35, 1,40, 1,34, 1,32, 1,30, 1,46, 1,39, 1,40 e 1,32 Å, respectivamente, para a molécula adsorvida, possivelmente porque O 2 é um aceitador de elétrons. Além disso, o comprimento da ligação de TM – O na maioria O 2 - Os sistemas (TM-fosforeno) são curtos devido à interação entre O 2 e os átomos de TM. O comprimento da ligação varia de 1,84 a 2,19 Å e resulta na formação de ligações químicas. Em particular, a ligação O – O é alongada para 1,40 Å, o valor mais alto entre os sistemas, no O 2 adsorvido molécula do sistema Pt-fosforeno. Assim, o sistema Pt-fosforeno é bastante adequado como um catalisador para a oxidação do CO, pois provavelmente possui alta capacidade catalítica.
A fim de obter mais informações sobre o mecanismo subjacente da alta atividade desses sistemas, selecionamos O 2 - (Pt-fosforeno) como exemplo e investigou sua densidade local de estados (LDOS). A Figura 4a mostra o LDOS projetado nos orbitais d de Pt no sistema Pt-fosforeno, orbitais d de Pt no O 2 - (Pt-fosforeno) sistema, a ligação O – O no O 2 - sistema (Pt-fosforeno), e a fase gasosa O 2 . No painel superior da Fig. 4a, um pico pode ser visto em E F - 0,6 eV, que se origina do orbital d parcialmente ocupado de Pt no sistema Pt-fosforeno. Esses estados devem ser responsáveis pela alta atividade do sistema Pt-fosforeno. Após a adsorção de um O 2 molécula, o LDOS projetado nos orbitais d de Pt abaixo do nível de Fermi é reduzido após a adsorção do O 2 devido à transferência de carga e aos estados acima do nível de Fermi também é substancialmente aumentado. Enquanto isso, o LDOS foi projetado no O 2 adsorvido molécula indica que o O 2 2 π * orbitais (orbital molecular mais baixo não ocupado, LUMO) estão se tornando parcialmente ocupados, o que diminuiu de seu valor de gás de E F + 2 eV para E F - 0,1 eV. Para esclarecimento, a diferença de densidade de carga do O 2 - O sistema (Pt-fosforeno) também é apresentado.
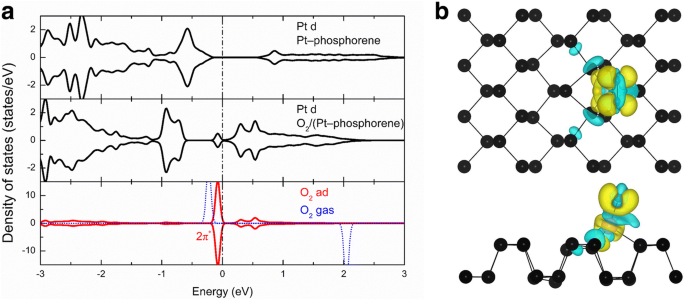
a Densidade local de estados (LDOS) de Pt e O 2 moléculas em Pt-fosforeno e O 2 -Sistemas de fosforeno-Pt e fase gasosa O 2 , respectivamente. b Diferença de densidade de carga no O 2 - sistema (Pt-fosforeno); a região amarela (ou seja, + 0,002 e / Å 3 ) e a região ciano (ou seja, - 0,002 e / Å 3 ) correspondem ao aumento e à perda, respectivamente, da densidade do elétron
A diferença de densidade de carga é definida da seguinte forma:
$$ {\ varDelta} _ {\ rho} ={\ rho} _T - {\ rho} _ {\ mathrm {molécula}} - {\ rho} _ {\ mathrm {absorbed}} $$ (3)
onde ρ T , ρ molécula e ρ absorvido são as cobranças totais no O 2 - sistema (Pt-fosforeno), O 2 molécula, e o sistema Pt-fosforeno, respectivamente. Como mostrado na Fig. 4b, a grande região amarela localizada no O 2 molécula indica que há uma transferência significativa de elétrons de Pt-fosforeno para O 2 , que também indica a forte hibridização orbital entre O 2 e o sistema Pt-fosforeno. De acordo com a análise de carga de Bader [54,55,56], 0,19 | e | é transferido do sistema Pt-fosforeno para o O 2 molécula. Portanto, a grande transferência de carga preenche os estados anti-ligação do O 2 molécula e enfraquece significativamente a ligação O – O. Da mesma forma, o mecanismo subjacente da alta atividade de outros sistemas também pode ser compreendido pela transferência de carga entre o O 2 molécula e o sistema TM-fosforeno. De fato, a análise de carga de Bader [54,55,56] mostrou que cargas de - 0,68, - 0,50, - 0,42, - 0,52, - 0,46, - 0,24, - 0,24, - 0,37, - 0,53, - 0,25, - 0,19 e - 0,09 | e | são transferidos do TM-fosforeno para a molécula de oxigênio no O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ru-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Pd-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Os-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Pt-fosforeno) e O 2 - (Au-fosforeno) sistemas, respectivamente.
Finalmente, estudamos as propriedades magnéticas de O 2 - Sistemas (TM-fosforeno). Os momentos magnéticos do O 2 - Os sistemas (TM-fosforeno) são mostrados na Tabela 3. O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno) e O 2 - Os sistemas (Ir-fosforeno) têm momentos magnéticos de 2,00, 1,00, 1,00, 1,14 e 1,00 μ B , respectivamente, que resultam da adsorção de um O 2 paramagnético molécula. A densidade de carga polarizada por spin destes O 2 - Os sistemas (TM-fosforeno) são exibidos na Fig. 5. Para o O 2 - (Fe-fosforeno) e O 2 - Sistemas (Co-fosforeno), acredita-se que o momento magnético surja principalmente do átomo do metal de transição e do O 2 molécula. Pelo contrário, para o O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Ir-fosforeno) e O 2 - Sistemas (Au-fosforeno), o momento magnético vem principalmente do O 2 molécula. Essas hipóteses são consistentes com os resultados apresentados na Tabela 3. Para melhor compreender como a adsorção de uma molécula de gás afeta a estrutura eletrônica do O 2 - Sistema (TM-fosforeno), as estruturas de banda eletrônica de cada sistema foram calculadas e os resultados são mostrados na Fig. 5. Primeiro, descobrimos que uma banda plana ocorre em torno do nível de Fermi ( E F ) após a adsorção de O 2 molécula em todos os sistemas, principalmente do O 2 molécula. Para o O 2 - (Fe-fosforeno), O 2 - (Co-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Ag-fosforeno) e O 2 - Sistemas (Au-fosforeno), os canais para divisão spin-up e spin-down revelam as características magnéticas. O 2 - (Fe-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Ir-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno) e O 2 - (Au-fosforeno) exibe comportamento semicondutor magnético, com um intervalo de banda considerável, exceto para o O 2 - Sistema (Co-fosforeno), que se revelou semimetálico. Esses resultados sugerem que os sistemas têm potencial para aplicação em spintrônica à base de fosforeno.
Densidades de spin do a O 2 - (Fe-fosforeno), b O 2 - (Co-fosforeno), c O 2 - (Ni-fosforeno), d O 2 - (Cu-fosforeno), e O 2 - (Ir-fosforeno), f O 2 - (Rh-fosforeno), g O 2 - (Ag-fosforeno) e h O 2 - Os sistemas (Au-fosforeno) são mostrados na linha superior; a estrutura de banda correspondente de cada sistema é mostrada na linha inferior. Na linha superior, um gráfico da densidade de carga polarizada por spin com um valor de iso-superfície de densidade de carga de 0,002 e / Å 3 é sobreposto nas vistas superior e lateral da estrutura cristalina do fosforeno puro; as regiões amarela e ciano correspondem a spins para cima e para baixo, respectivamente. Nos gráficos de estruturas de banda, as linhas pretas e vermelhas denotam canais de spin-up e spin-down, respectivamente; o nível de Fermi é definido como zero e é indicado pela linha tracejada cinza
Conclusões
Nós investigamos as propriedades estruturais, eletrônicas e magnéticas de diferentes sistemas de fosforeno-TM. Todos os adátomos preferiram ocupar o sítio oco no fosforeno. A energia de adsorção considerável revela que todos os sistemas de adsorção de fosforeno-TM são bastante robustos, indicando que o fosforeno forma ligações fortes com todos os 12 tipos de adátomos TM. Além disso, descobrimos que a dopagem com Fe, Co e Au pode resultar em propriedades semicondutoras magnéticas em fosforeno monocamada, com momentos magnéticos totais de 2, 1 e 0,96 μ B , respectivamente.
Além disso, também examinamos as propriedades de um O 2 molécula adsorvida no sistema TM-fosforeno. Foi muito encorajador descobrir que todos os O 2 - Sistemas (TM-fosforeno), exceto para O 2 - (Pd-fosforeno), apresentam boa atividade catalítica para a oxidação do CO devido ao alongamento da ligação O – O. O 2 - (Fe-fosforeno), O 2 - (Ni-fosforeno), O 2 - (Cu-fosforeno), O 2 - (Rh-fosforeno), O 2 - (Ag-fosforeno), O 2 - (Ir-fosforeno) e O 2 - Os sistemas (Au-fosforeno) exibem propriedades semicondutoras polarizadas de spin com momentos magnéticos de 2,00, 2,00, 1,00, 1,00, 1,14, 1,00 e 1,00 μ B . O 2 - (Co-fosforeno) apresenta características semi-metálicas magnéticas, com um momento magnético de 2,00 μ B . Portanto, nossos resultados podem abrir novas possibilidades para a aplicação de fosforeno nas áreas de catálise e spintrônica.
Abreviações
- 2D:
-
Bidimensional
- B:
-
Ponte
- GGA:
-
Aproximação de gradiente generalizado
- H:
-
Site oco
- LDOS:
-
Densidade local de estados
- PBE:
-
Perdew-Burke-Ernzerh
- T:
-
Topo de um átomo de fósforo
- TM:
-
Metal de transição
Célula solar híbrida de silício orgânico nanoestruturado de alto desempenho com estrutura de superfície modificada
Síntese e estudo in vitro de uma sonda de modo duplo que direciona a integrina αvβ3
Nanomateriais
- Construção do Relé
- Eletromagnetismo
- Videotape
- Ímã
- Disquete
- Tipos de magnetômetros
- Nanodiamonds para sensores magnéticos
- Nanocluster para conduzir plasmons magnéticos
- Magnetismo de Percolação em Nanopartículas Ferroelétricas
- Estudo de primeiros princípios sobre a estabilidade e imagem STM de Borophene