


Propriedades eletrônicas da poltrona de nanoribbons de fósforo preto modificada por elementos de transição V, Cr e Mn
Resumo
As propriedades estruturais, elétricas e magnéticas de nanofitas de fosforeno preto de poltrona (APNRs) funcionalizadas por borda por elementos metálicos de transição (TM) V, Cr e Mn foram estudadas pela teoria do funcional de densidade combinada com a função de Green fora de equilíbrio. Os estados de borda spin-polarizados introduzem uma grande variedade nas estruturas eletrônicas dos TM-APNRs. Para APNRs com borda costurada em Mn, suas estruturas de banda exibem propriedades elétricas semicondutoras no estado ferromagnético. Um campo elétrico transversal pode então tornar os Mn-APNRs metálicos, deslocando as bandas de condução dos estados de borda por meio do efeito Stark. A heterojunção Mn / Cr-APNR pode ser usada para fabricar spin p-n diodo onde a retificação forte atua apenas em um spin.
Introdução
A descoberta do grafeno [1, 2] desencadeou uma onda de pesquisas para materiais de cristal bidimensional (2D) [3,4,5,6]. Na última década, nitreto de boro hexagonal, dichalcogeneto de metal de transição, fosforeno preto e muitos outros foram preparados ou previstos [7,8,9]. Esses materiais 2D podem ser implementados em uma ampla gama de campos, importantes não apenas para explorar novos fenômenos físicos e desempenho sob o limite 2D, mas também para muitas novas aplicações em dispositivos eletrônicos, spintrônicos e optoeletrônicos [10,11,12,13 , 14,15,16,17,18,19,20,21]. Além disso, algumas propriedades de materiais bidimensionais podem ser melhoradas após serem adaptadas em nanofitas unidimensionais (1D) ou / e funcionalizadas [22, 23]. Excelente desempenho foi observado em transistores de efeito de campo de nanofibra de grafeno sintetizado de baixo para cima [24]. Contatos sem barreira de Schottky com semicondutores 2D via carboneto de metal ou nitreto funcionalizado por grupos O ou OH foram previstos [25]. Nanoflocos de fósforo modificados por borda foram propostos para células solares altamente eficientes [26]. Defeitos atômicos e impurezas podem ser empregados para modular localmente as propriedades eletrônicas para aplicações potenciais em magnetismo e catálise [27,28,29]. A aplicação de campo elétrico externo e heteroestruturas pode ainda manipular significativamente as propriedades eletrônicas [30,31,32].
Entre os materiais 2D conhecidos, o fosforeno preto é um dos poucos com propriedades mecânicas, elétricas e ópticas superiores para aplicações em dispositivos. Desde a fabricação de transistores de efeito de campo baseados nele [9], o fosforeno preto tem atraído cada vez mais interesse. É um semicondutor direto com intervalo de banda modesto (≈ 2 eV) e alta mobilidade de orifício (≈ 1000 cm 2 / (Vs)) [33,34,35], mostrando um enorme potencial de aplicação nas áreas de eletrônica, optoeletrônica, sensores, catálise e baterias [36,37,38,39]. Semelhante ao grafeno, o fosforeno preto pode ser cortado ao longo de duas direções típicas em nanofitas de fosforeno em zigue-zague (ZPNRs) ou nanofitas de fosforeno de poltrona (APNRs) [40,41,42]. A simulação dos primeiros princípios mostrou que a dopagem substitucional do metal de transição pode facilmente introduzir magnetismo no fosforeno para aplicações spintrônicas [43]. A absorção de metais de transição, ancorada por defeitos, pode dar origem a sistemas de fosforeno compostos semi-metálicos e metálicos [44]. Foi previsto que a modificação da borda do metal de transição também pode modular muito as propriedades eletrônicas das nanofitas de fosforeno em zigue-zague [45]. No entanto, até onde sabemos, os efeitos da passivação da MT nos APNRs ainda não foram bem estudados.
Neste artigo, enfocamos a modulação das propriedades eletrônicas de APNRs funcionalizados por elementos metálicos de transição típicos V, Cr e Mn, uma vez que eles apresentam momentos magnéticos maiores do que os demais. As simulações baseadas na teoria do funcional da densidade mostram que o comportamento do semicondutor pode aparecer e ser controlado por um campo elétrico transversal. Além disso, spin de alto desempenho p-n junção pode ser projetada para aplicações spintrônicas [46].
Sistemas e métodos computacionais
O fósforo preto é um material em camadas em que as camadas atômicas são empilhadas juntas por uma fraca força de van der Waals entre camadas, enquanto os átomos em cada camada são ligados por fortes ligações covalentes. Pode ser facilmente descascado em fosforenos de monocamada. A vista superior de um fosforeno é esquematizada na Fig. 1a com uma parte ampliada em seu lado direito para mostrar os parâmetros de geometria. Duas vistas laterais ao longo das direções da poltrona e do zigue-zague, respectivamente, são fornecidas além. Cada átomo de fósforo está ligado a três átomos de fósforo adjacentes (com constantes de rede 3,31 e 4,38 Å, comprimento de ligação 2,2 Å, ângulo de ligação 96,34 ° e ângulo diédrico de 102,1 °) para formar uma estrutura em colmeia plissada [47]. Como outros materiais bidimensionais de rede de favo de mel hexagonal, como grafeno e dissulfeto de molibdênio, um fosforeno pode ser adaptado para nanofitas com duas morfologias de borda típicas, a poltrona e nanofitas de fosforeno preto em zigue-zague [40, 41, 48, 49].
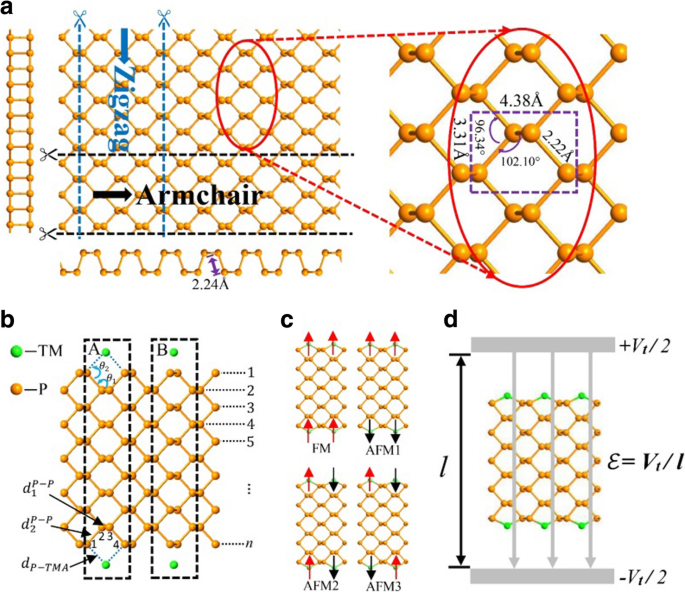
a Vistas superior e lateral de um fósforo 2D com uma visão ampliada no lado direito. As vistas em corte transversal da poltrona e bordas em zigue-zague são mostradas abaixo e no lado esquerdo, respectivamente. b Um APNR com adátomos TM em locais ocos (A) e locais superiores (B) na borda. Os quadros tracejados indicam o tamanho da célula primitiva e o número n denota a largura do nanoribão. c As quatro configurações magnéticas dos APNRs. d O diagrama esquemático na presença de um campo elétrico transversal
Aqui, consideramos o semicondutor n -APNRs para número de largura ímpar n com seção transversal simétrica espelhada. Devem seguir-se resultados semelhantes para n mesmo uma vez que as duas arestas do nanofibra são quase independentes, conforme abordado a seguir. Os efeitos das modificações de borda por três elementos V, Cr e Mn típicos de metal de transição (TM) são analisados sistematicamente. Conforme ilustrado na Fig. 1b, um átomo de TM pode ser adsorvido a uma aresta APNR na posição oca (caso A) ou na posição superior (caso B). Como o caso A tem uma energia de ligação muito maior, nós o adotamos onde um átomo de TM é adsorvido próximo ao centro de cada posição oca e se liga aos dois átomos da borda de fósforo. Para facilitar a descrição da geometria de ligação dos átomos de TM nas bordas do APNR, como ilustrado na Fig. 1b, denotamos os átomos de fósforo nos locais 1, 2, 3 e 4 como P 1 , P 2 , P 3 , e P 4 , respectivamente. Definimos também alguns parâmetros geométricos:os comprimentos de ligação \ ({d} _1 ^ {P-P} \) (entre P 2 e P 3 ), \ ({d} _2 ^ {P-P} \) (entre P 1 , P 2 ou P 3 , P 4 ) e d P - TM e os ângulos de ligação θ 1 (entre \ ({d} _1 ^ {P-P} \) e \ ({d} _2 ^ {P-P} \)) e θ 2 (entre \ ({d} _2 ^ {P-P} \) e d P - TM ) Devido ao magnetismo dos adátomos TM, existem quatro configurações magnéticas possíveis, ou seja, FM, AFM1, AFM2 e AFM3, conforme mostrado na Fig. 1c. Na ausência de campo magnético, nossa simulação mostra que a energia da célula unitária AFM2 na Fig. 1c é cerca de 0,2 eV menor do que a da FM. As duas arestas são quase independentes e a polarização de spin oposta entre elas nas configurações AFM1 e AFM3 pode reduzir a energia em uma quantidade inferior a 0,002 eV. Neste artigo, estudamos as propriedades eletrônicas das nanofitas na configuração FM, uma vez que um campo magnético aplicado pode mantê-las assim. Estudamos também os efeitos de um campo elétrico transversal aplicado, conforme ilustrado na Fig. 1d, na estrutura eletrônica e nas propriedades de FM APNRs. Finalmente, propomos possíveis aplicações de dispositivos dos materiais.
As propriedades de transporte de uma junção de nanofibra são calculadas estabelecendo uma estrutura de dispositivo de duas sondas. A junção é dividida em três partes:Uma região de espalhamento, onde a interface da junção está localizada, é imprensada entre os eletrodos esquerdo (L) e direito (R). Quando uma polarização de tensão V b é aplicado entre os dois eletrodos, definimos as energias de Fermi nos eletrodos L e R como μ L =- e | V b | / 2 e μ R = e | V b | / 2. A corrente eletrônica de spin σ através dos dispositivos quânticos é avaliada pela fórmula de Landauer-Büttiker [50]:
$$ {I} _ {\ sigma} =\ frac {e} {h} \ underset {- \ infty} {\ overset {\ infty} {\ int}} {T} _ {\ sigma} (E) \ esquerda [f \ esquerda (E - {\ mu} _ {\ mathrm {R}} \ direita) -f \ esquerda (E - {\ mu} _ {\ mathrm {L}} \ direita) \ direita] dE $ $ (1)
Aqui, T σ ( E ) é a transmissão de spin σ e f a função de distribuição de Fermi-Dirac.
A simulação é realizada pelo pacote de kits de ferramentas Atomistix (ATK) com base na teoria do funcional de densidade ab initio (DFT) combinada com o método da função de Green sem equilíbrio (NEGF) [51, 52]. Antes da estrutura eletrônica e das simulações de transporte, as estruturas são otimizadas até que as forças atuantes em cada átomo sejam menores que 0,02 eV / Å. Usamos a aproximação de gradiente generalizado dependente de spin com a parametrização de Perdew-Burke-Emzerhof (SGGA-PBE) para o funcional de correlação de troca. Confirmamos que as simulações SGGA + U levam ao mesmo resultado apresentado a seguir [43]. Um conjunto básico de polarização zeta dupla ( dzp ) orbitais atômicos é usado no cálculo para obter um resultado preciso. Uma camada de vácuo de 20 Å de espessura é inserida entre as nanofitas vizinhas para evitar acoplamentos entre fitas. A energia de truncamento para a expansão do vetor de base das funções de onda é definida como 150 Hartree ou 4082 eV com um k -Space mesh grid de 1 × 1 × 101. Uma temperatura eletrônica de 300 K é adotada na técnica de integração do eixo real para o esquema NEGF para facilitar a simulação. As quatro configurações magnéticas são obtidas definindo inicialmente as polarizações de spin correspondentes dos adátomos TM antes da otimização. O campo elétrico transversal ε é gerado por duas placas de metal virtuais paralelas, separadas por uma distância l , com uma diferença de potencial elétrico V t então ε =V t / l .
Resultados e discussão
Geometria e energia de ligação
Em APNRs primitivos, os átomos P da borda mudam para a posição oca, de modo que cada "poltrona" da borda se torna mais estreita em comparação com sua contraparte 2D, como mostrado nas Fig. 2a, b. Se um APNR é hidrogenado com a ligação em suspensão de cada átomo P da borda saturado por um átomo de H, conforme abordado na Ref. [48, 53], os átomos P da borda se recuperam para suas posições 2D, conforme ilustrado na Fig. 2c. Quando um átomo de TM é adsorvido em cada posição oca, ele passiva os dois átomos P da aresta. As poltronas então se recuperam parcialmente e as bordas tornam-se magnetizadas devido à polarização do spin dos adátomos TM. Na configuração FM, nenhuma reconstrução é observada nas bordas e o comprimento da célula primitiva permanece inalterado, conforme indicado na Fig. 2d-f.
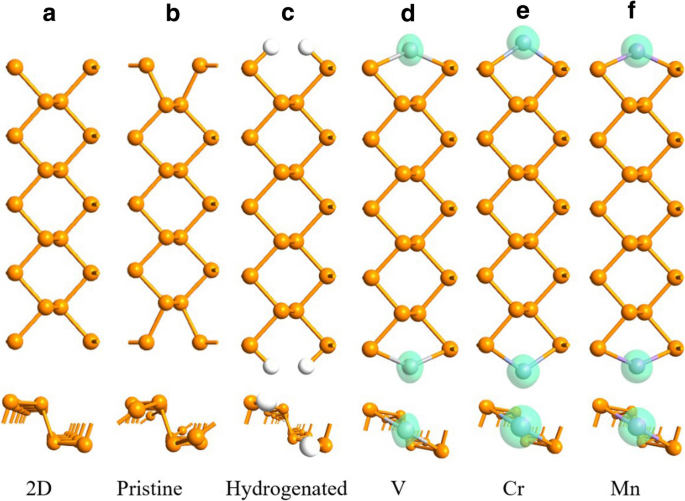
Geometrias de FM 9-APNRs a acabado de cortar de um fosforeno 2D, b geometricamente otimizado (puro), c hidrogenado, e após adsorver d V, e Cr e f Mn átomos no limite. A densidade de polarização de spin dos átomos é mostrada pela isosuperfície verde no valor de 0,004 e / Å 3
Na Tabela 1, listamos os parâmetros de geometria e a energia de ligação E b para 9- e 17-APNRs puros, hidrogenados e com adsorção de TM na configuração FM, se aplicável. Aqui, E b =( mE X + E APNR - E X - APNR ) / m com E X , E APNR , e E X - APNR as energias totais de um átomo externo, uma célula primitiva de APNR primitivo e uma célula primitiva de APNR passivada por m átomos externos, respectivamente, com m =4 para H e m =2 para elementos TM. Quando cortamos um fosforeno 2D para fazer um APNR, as ligações de suspensão na borda reduzem significativamente θ 1 de 102 a 87 °. A passivação das ligações em suspensão por átomos externos recupera θ 1 e introduz uma reação repulsiva, marcada pelo trecho de \ ({d} _ {P-P} ^ 1 \) e \ ({d} _ {P-P} ^ 2 \). Nos casos de TM, a adsorção de átomos de V mostra a reação repulsiva mais forte com o maior θ 1 . Semelhante ao de H, a adsorção do elemento TM é energeticamente estável com uma energia de ligação da ordem de 4 eV. As duas arestas dos APNRs são quase independentes uma da outra, então os parâmetros de geometria e E b são insensíveis à largura do APNR. A geometria de ligação e a energia também se mantêm em diferentes configurações magnéticas para TM- n -APNRs.
Estrutura eletrônica e propriedades magnéticas
Na Fig. 3, apresentamos as estruturas de banda e funções de onda típicas de elétrons em 9-APNRs com e sem modificação de borda. APNRs primitivos são semicondutores indiretos não magnéticos com um gap de E g ≈ 0,5 eV, em que os estados eletrônicos na parte superior (inferior) da banda de valência (condução) são estados em massa (borda). Quando os átomos P da borda são passivados por átomos de H, a banda de condução devido às ligações de suspensão da borda em APNRs primitivos se afasta do gap e os ANPRs hidrogenados tornam-se semicondutores diretos com um gap maior de E g ≈ 1,0 eV. Os estados na parte inferior da banda de condução e no topo da banda de valência são todos estados em massa. Conforme a largura aumenta de n =9 a 17, o gap diminui ligeiramente de 1,01 para 0,89 eV de acordo com aqueles previstos por Han et al. [49].
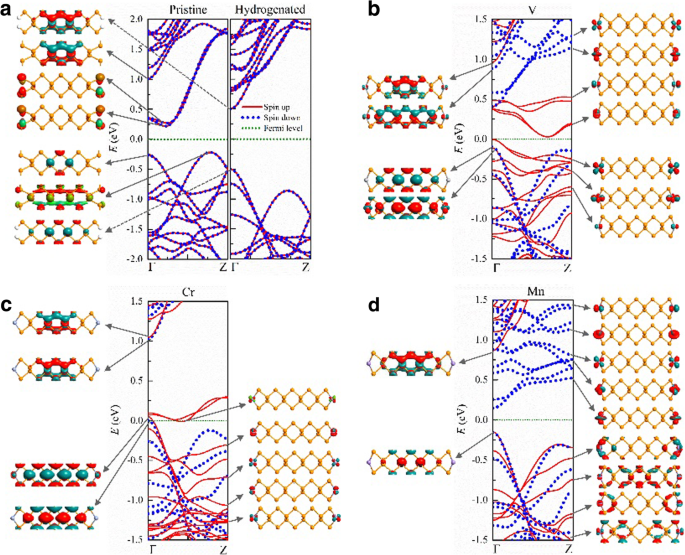
Estruturas de banda e funções de onda típicas perto da energia de Fermi de 9-APNRs primitivos modificados por a H, b V, c Cr e d Átomos Mn
Quando os átomos de TM são adsorvidos nas bordas dos APNRs, eles permanecem polarizados por rotação. Na configuração FM, V- n -APNRs são semicondutores magnéticos com gap dependente de spin. Conforme ilustrado na Fig. 3b, para n =9, os elétrons de spin para cima têm uma lacuna indireta de \ ({E} _g ^ {\ mathrm {up}} \ aproximadamente 0,03 \) eV, enquanto os elétrons de spin para baixo têm uma lacuna direta de \ ({E} _g ^ {\ mathrm {baixo}} \ aprox 0,5 \) eV. Os estados eletrônicos nas bandas de spin-up em torno da energia de Fermi são compostos por d orbitais dos V adátomos e são confinados nas bordas. Essas bandas de borda de spin-up têm dispersão semelhante e estão parcialmente ocupadas. A parte superior da banda de valência correspondente e a parte inferior da banda de condução se separam no k espaço, mas estão próximos uns dos outros em energia. Uma estreita lacuna de banda indireta aparece para elétrons de spin-up. Em contraste, todas as bandas de borda de spin-down estão muito acima da energia de Fermi. A banda de valência de spin-down é de estados de massa e tem dispersão oposta da banda de condução de spin-down, que é de estados de borda. Isso resulta no gap direto para elétrons de spin-down. As bandas da borda V aparecem em pares devido ao fraco acoplamento entre os átomos V das bordas esquerda e direita. Três dos cinco pares estão ocupados, então cada célula primitiva tem um momento magnético de 6 μ B .
Um par de spin-up e todo o spin-down d bandas de borda orbital estão localizadas acima do nível de Fermi em Cr-9-APNR como ilustrado na Fig. 3c, porque há quatro d elétrons orbitais em cada átomo de Cr. Devido à ligeira sobreposição dos dois pares mais altos de bandas de borda spin-up perto do topo da banda de valência spin-down, ele se torna um meio metal com o nível de Fermi logo acima do topo da banda de valência spin-down. Em Mn-9-APNR, todos os cinco pares de spin-up d bandas orbitais são ocupadas enquanto o spin-down d as bandas orbitais estão vazias como mostrado na Fig. 3d. Torna-se um meio semicondutor onde as lacunas de banda de spins opostos diferem muito, com \ ({E} _g ^ {\ mathrm {up}} \ approx 1 \) eV para spin-up e \ ({E} _g ^ {\ mathrm {down}} \ approx 0.3 \) eV para spin-down. Ambos os spins têm o mesmo topo da banda de valência, na qual estão os estados em massa. No entanto, o fundo da banda de condução do spin-down é muito menor do que o spin-up devido aos estados de borda do spin-down desocupados.
As estruturas eletrônicas de TM- n -APNRs permanecem o mesmo padrão e não mudam tanto quanto n aumenta conforme ilustrado na Fig. 4. No entanto, as propriedades físicas podem variar significativamente nas amostras passivadas de Cr porque uma lacuna de energia pode abrir como n aumenta. Narrow Cr- n -APNRs são meio metal, mas amplo Cr- n -APNRs podem se tornar semicondutores, conforme mostrado nas inserções da Fig. 4 para n =11 e n =17, respectivamente.
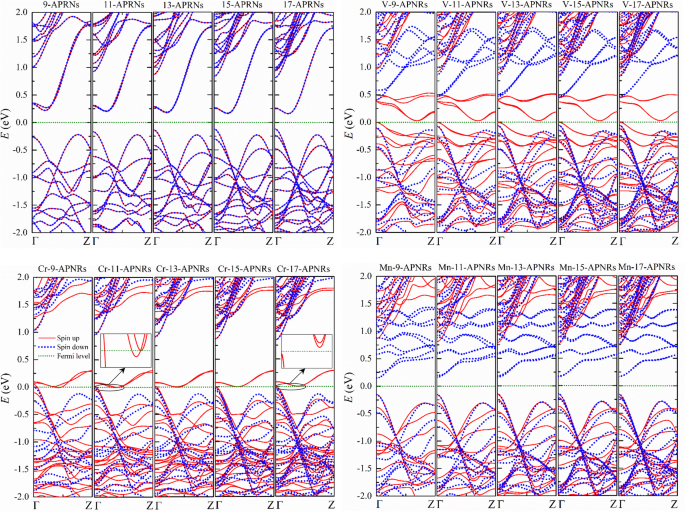
Estruturas de bandas de n primitivas -APNRs e aqueles modificados por borda por átomos V, Cr e Mn para vários n . Visualizações ampliadas de Cr- n -APNRs próximos ao nível de Fermi são mostrados nas inserções para n =11 e 17
Os perfis de distribuição de momento magnético de FM TM-9-APNRs são mostrados na Fig. 2, onde as isosuperfícies da densidade de spin ∆ρ = ρ up - ρ baixo =0,004 e / Å 3 são plotados. Aqui, ρ up e ρ baixo são as densidades de elétrons de spin up e spin down, respectivamente. Os momentos magnéticos estão concentrados principalmente em torno dos átomos de TM, e a contribuição dos átomos de P é muito pequena para ser mostrada claramente. Na Tabela 2, apresentamos o momento magnético total M T em uma célula primitiva, a soma dos momentos dos dez átomos da aresta M E =2 M (TM) + 4 M (P 1 ) + 4 M (P 2 ), e o momento de um átomo de aresta única TM, P 1 / P 4 ou P 2 / P 3 .
Os momentos magnéticos totais vêm principalmente dos átomos da borda ( M T ≈ M E ) e em unidade de μ B por célula primitiva estão próximos do número de elétrons de valência dos átomos do metal de transição menos 4. Em V- n -APNRs, os átomos P da borda (P 1 e P 4 ) são polarizados antiparalelamente ligeiramente, enquanto os átomos P da segunda aresta (P 2 e P 3 ) são polarizados paralelamente. Assim, os momentos magnéticos dos átomos P são quase cancelados entre si. Cada átomo de V tem um momento magnético de cerca de 3 μ B de três 3 d orbitais. Os 4 s orbital está totalmente ocupado semelhante a um único átomo de V. Em contraste, os átomos P da aresta em Cr- n -APNRs têm momentos magnéticos muito maiores de M (P1) ≈ - 0,27 μ B . Coincidentemente, eles têm o d mais longo P - TM entre os três TM-APNRs, indicando também o maior desvio de geometria dos átomos P daqueles no fosforeno 2D. Além disso, cada átomo de Cr tem um momento magnético de aproximadamente 5 μ B , em vez de 4 μ B . Isso sugere que seus 4 s orbital não está totalmente ocupado e contribui para a polarização do spin, semelhante ao caso do átomo de Cr isolado com uma configuração de elétron de valência de 3 d 5 4 s 1 . Os s spin-polarizados orbitais de átomos de Cr em Cr-APNRs podem ter induzido a polarização de spin antiparalelo no p orbitais em seus átomos P vizinhos por meio do mecanismo de troca cinética. Em Mn- n -APNR, o d orbitais do átomo de Mn são ocupados pela metade com um momento magnético de cerca de 5 μ B e os átomos P vizinhos são polarizados paralelamente muito fracamente. Na Fig. 5, representamos graficamente a densidade parcial de estados (PDOS) (azul) de d orbitais em átomos de TM junto com a densidade total de estados (DOS) (preto) de 9-APNRs. Aqui, a divisão do spin e a propagação da energia de d orbitais são mostrados claramente. Em APNRs primitivos e hidrogenados, os espectros de DOS de spin-up e spin-down se sobrepõem, indicando que não há polarização de spin. Em TM-APNRs, a rotação para cima e para baixo d Os espectros orbitais de PDOS se distribuem principalmente em uma faixa de energia de 2 a 4 eV. Eles estão bem separados em energia com uma separação de cerca de 3, 9 e 4 eV em V-, Cr- e Mn-APNR, respectivamente. Excluindo o d orbitais, o p orbitais de átomos de P dominam a contribuição para o DOS das bandas de valência. Observe que o s orbitais de átomos de Cr também contribuem significativamente em Cr-APNRs. A passivação de borda de átomos de Co e Ni também pode introduzir magnetismo em APNRs, mas o magnetismo introduzido por outros elementos TM como Sc, Ti, Fe, Cu e Zn pode ser bastante limitado.
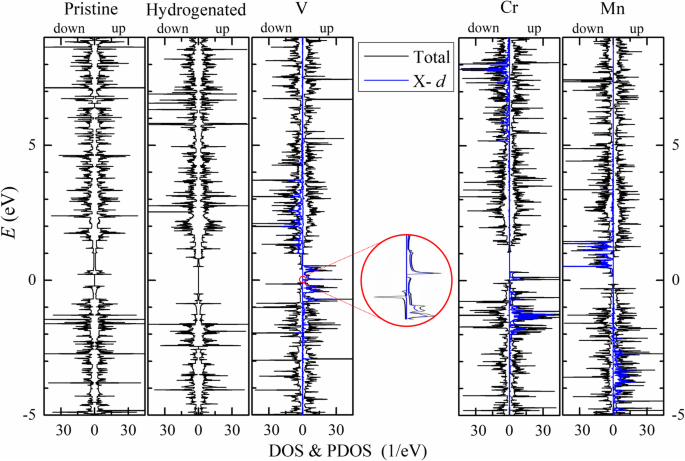
O DOS (curva preta) de 9-APNRs originais e modificados em seu estado FM é traçado para o spin para cima (direita) e para baixo (esquerda). O d PDOS orbital (curva azul) de átomos de TM também é apresentado para comparação. O DOS do V-9-APNR perto da energia Fermi é ampliado para mostrar o intervalo de banda
Efeitos de um campo elétrico transversal
O campo elétrico transversal tem sido amplamente empregado em dispositivos eletrônicos para controlar a concentração de portadores e a estrutura de banda de semicondutores [54, 55]. Conforme indicado na Fig. 1d, simulamos as estruturas eletrônicas de TM- n -APNRs na configuração FM sob um campo elétrico transversal \ (\ mathcal {E} ={V} _t / l \) paralelo ao plano de nanofita, via sanduichamento de nanofitas entre duas barras paralelas. Aqui, V t é a diferença de tensão entre as duas barras e l é a separação entre eles. Devido ao efeito Stark, dois estados degenerados separados no espaço real por uma distância Δ ao longo do campo elétrico devem se dividir por uma quantidade de \ (\ delta E =e {\ mathcal {E}} ^ {\ ast} \ Delta \) , onde o campo elétrico efetivo \ ({\ mathcal {E}} ^ {\ ast} \) é geralmente menor do que o campo elétrico externo \ (\ mathcal {E} \) como resultado do efeito de blindagem. Em TM- n -APNRs, a distância Δ entre os centros de estado de um par de banda de borda pode ser tanto quanto a largura do nanofibra se cada estado for confinado apenas em uma borda, mas Δ deve ser mais curto ou mesmo desaparecer para estados de borda mistos. Conforme ilustrado pelas funções de onda na Fig. 3, os estados das bordas geralmente são misturados.
Na Fig. 6, apresentamos as estruturas de banda de V-, Cr- e Mn-13-APNR para vários \ (\ mathcal {E} \). A largura da nanofita é de cerca de \ (w =0,5 \ left (n-1 \ right) \ times 3,31 \ {\ AA} + {d} ^ {P- \ mathrm {TM}} \ cos \ left ({135} ^ {{} ^ {\ circ}} - {\ theta} _2 \ right) \ approx 21 \ kern0.20em {\ AA}. \) A divisão Stark é muito menor que \ (e \ mathcal {E} w \) indicando forte efeito de triagem ou forte mistura dos estados de borda. Como o V-13-APNR tem uma lacuna de banda de spin-up muito estreita, ele se torna meio metálico em cerca de \ (\ mathcal {E} =3 \) V / nm. A divisão Stark das bandas da borda de condução pode chegar a 0,1 eV em \ (\ mathcal {E} =5 \) V / nm. O Cr-13-APNR mostra uma força semelhante à divisão Stark e permanece meio metálico sob o campo transversal.
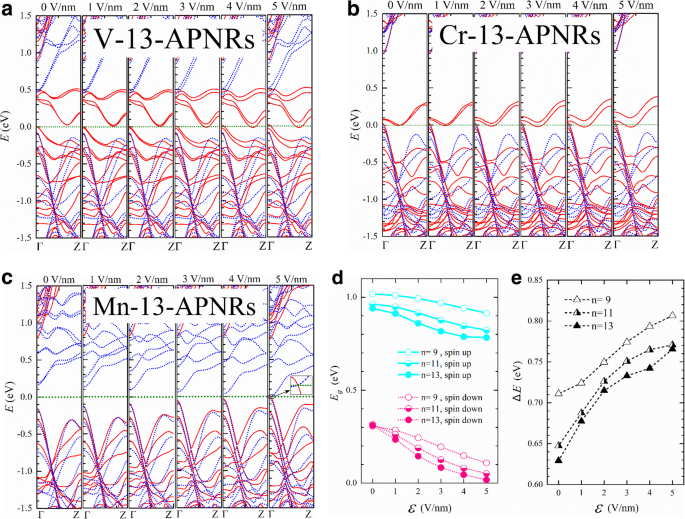
As estruturas de banda de spin-up (sólido) e spin-down (pontilhado) de a V-, b Cr- e c Mn-13-APNRs sob um campo elétrico transversal de força \ (\ mathcal {E} =0,1, \ dots, 5 \) V / nm. d Os intervalos de banda de Mn- n -APNRs versus \ (\ mathcal {E} \) para rotação para cima (\ ({E} _g ^ {\ mathrm {para cima}} \), linhas sólidas) e rotação para baixo (\ ({E} _g ^ {\ mathrm {down}} \), linhas pontilhadas) com n =9, 11 e 13. e A diferença de intervalo \ (\ Delta E ={E} _g ^ {\ mathrm {para cima}} - {E} _g ^ {\ mathrm {para baixo}} \) versus \ (\ mathcal {E} \)
Um efeito Stark muito mais forte é observado no semicondutor Mn-13-APNR como mostrado na Fig. 6c. Os pares de bandas de condução de spin-down de estados de borda obtêm uma divisão de cerca de 0,55 eV no ponto Γ em k espaço sob \ (\ mathcal {E} =5 \) V / nm. A banda de condução spin-down se sobrepõe à banda de valência spin-up, e o Mn-13-APNR transita de um semicondutor para um metal, conforme ilustrado pela inserção ampliada. Na Fig. 6d, representamos graficamente as lacunas de energia do spin-up e spin-down versus a intensidade do campo. As funções de onda do elétron mudam com o campo e as lacunas de energia não variam linearmente com o campo. O gap de Mn-13-APNRs quase desaparece em \ (\ mathcal {E} =5 \) V / nm para elétrons spin-down, mas permanece acima de 0,75 eV para elétrons spin-up. A diferença da lacuna de energia ∆E entre os spins opostos é plotado versus \ (\ mathcal {E} \) na Fig. 6e para n =9, 11 e 13. ∆E aumenta em uma etapa muito mais lenta para n =9 do que para n =11 e 13 no campo baixo, mas a maneira se inverte no campo alto.
Spin p-n Junção
Vimos que os átomos de TM podem modular a estrutura de banda dos APNRs de várias maneiras. Isso oferece oportunidades para um novo design de dispositivo. Por exemplo, podemos combinar Cr-APNRs e Mn-APNRs para formar um p-n dependente de spin junção. Experimentalmente, a dopagem com íons metálicos [56] em fosforeno está disponível. Costura suave de materiais 2D [57] e modificação de borda atômica de nanofitas também podem ser realizadas [58]. Essas técnicas podem ser usadas para fabricar o p-n junção. Na Fig. 7a, traçamos sua corrente-tensão ( I-V ) característica obtida da simulação do sistema de duas sondas mostrada no destaque superior. O giro p-n junção mostra efeito de retificação muito forte para elétrons de spin-up, mas apenas efeito fraco para elétrons de spin-down. Essa dependência de spin vem das estruturas de banda distintas dos eletrodos esquerdo e direito, conforme ilustrado na inserção inferior. Sob polarização negativa, o eletrodo Mn-APNR esquerdo tem uma energia de Fermi μ L = e | V b | / 2 e o Cr-APNR correto μ R =- e | V b | / 2. Dentro da janela de transporte da faixa de energia [ μ L , μ R ], há apenas uma parte muito pequena da banda de energia de spin-down no eletrodo Cr-APNR, então a corrente de spin-down permanece baixa. Em contraste, uma ampla sobreposição das bandas de energia de spin-up existe nos eletrodos Mn- e Cr-APNR e a corrente de spin-up aumenta rapidamente com a polarização. Dentro da janela de transporte [ μ R , μ L ] sob polarização positiva, no entanto, não há banda de energia de spin-up no eletrodo esquerdo e a corrente correspondente permanece quase zero, pois Mn-APNR é um p -tipo semicondutor de gap largo para aumento de rotação. A corrente de spin-down começa a aumentar em V b =0,2 V quando a energia de Fermi direita se alinha com a banda de condução de spin-down esquerda. Na Fig. 7b, traçamos a razão de retificação α σ =[ eu σ (- | V b |) - I σ (| V b |)] / I σ (| V b |) de rotação σ em função da magnitude do viés | V b | Em | V b | =0,5 V, o APNR spin p-n junção tem uma retificação de 2400 para aumento de spin e apenas 2 para down spin.
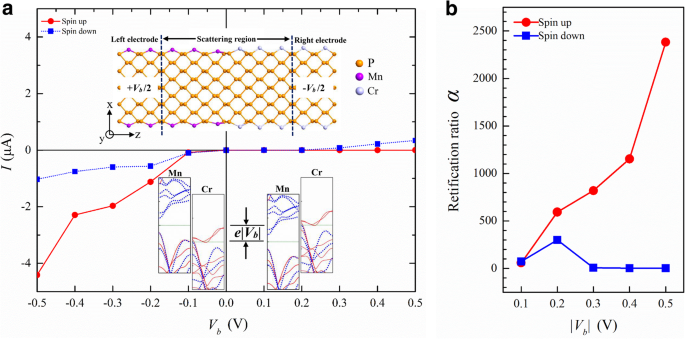
a O dependente de spin I-V característica de uma heterojunção Mn / Cr-9-APNR. A geometria do sistema de duas sondas é mostrada na inserção superior. As inserções inferiores esquematizam os alinhamentos das bandas de energia do eletrodo para polarizações negativas e positivas. b A taxa de retificação correspondente α é plotado versus a magnitude do viés
Conclusões
A simulação DFT-NEGF sugere que a funcionalização de borda de átomos de TM pode manipular muito as propriedades elétricas e magnéticas de APNRs semicondutores não magnéticos e torná-los metálicos ou semicondutores. Os átomos de TM em TM-APNRs mantêm suas configurações eletrônicas em estado isolado, onde o magnetismo dos átomos V e Mn vem principalmente de d orbitais, mas o de Cr de ambos d e s orbitais. Em Mn-APNRs, o d orbitais estão meio preenchidos. Todo o spin-up d orbitais dos átomos de Mn estão ocupados e o spin-down d orbitais estão acima do nível de Fermi. Devido ao estreito gap de d orbital, Mn-APNRs tornam-se semicondutores onde as bandas de energia de spin-down têm uma lacuna muito mais estreita no nível de Fermi do que as de spin-up. Esta propriedade peculiar pode ser empregada para o projeto de dispositivos spintrônicos, uma vez que os materiais podem ser semicondutores para um spin e isolante para o outro em condições adequadas. Com a ajuda do efeito Stark nos estados de borda, as lacunas de energia podem ser moduladas por um campo elétrico transversal aplicado. Por exemplo, um campo de 5 V / nm pode fechar o gap de elétrons de spin down enquanto mantém um gap de 0,75 eV para elétrons de spin up. Aproveitando a diferença drástica de banda de energia entre Mn- e Cr-APNRs, podemos projetar spin p-n diodos da junção Mn / Cr-APNR nos quais ocorre forte retificação em apenas um spin.
Abreviações
- 1D:
-
Unidimensional
- 2D:
-
Bidimensional
- AFM:
-
Antiferromagnético
- APNR:
-
Poltrona nanoribão de fosforeno preto
- ATK:
-
Kits de ferramentas Atomistix
- DFT:
-
Teoria da densidade funcional
- DOS:
-
Densidade de estados
- FM:
-
Ferromagnético
- NEGF:
-
Função de Green fora de equilíbrio
- TM:
-
Metal de transição
WO3 / p-Type-GR Camadas Materiais para Degradação Antibiótica Fotocatalítica Promovida e Dispositivo para Mecanismo Insight
Conversão de fotocondutividade positiva e negativa induzida por H2O adsorção de molécula em WO3 Nanowire
Nanomateriais
- Propriedades e usos do fluxo de tungstênio
- Propriedades e aplicações do tântalo
- Preparação e propriedades magnéticas de nanopartículas de espinélio FeMn2O4 dopadas com cobalto
- Em direção aos nanofluidos de TiO2 - Parte 1:Preparação e propriedades
- Estrutura e propriedades eletrônicas da nanoargila caulinita dopada com metal de transição
- Modulação das propriedades de anisotropia eletrônica e óptica de ML-GaS por campo elétrico vertical
- Estados eletrônicos do nanocristal dopado com oxigênio e emissão visível em silício negro preparado por ns-Laser
- Propriedades ópticas e eletrônicas de fotodiodos N + / P de silício hiperdopado com enxofre induzido por laser de femtosegundo
- Metal de cromo:elementos, propriedades e usos
- Propriedades de PCB automotivo e considerações de design