


Estrutura e propriedades eletrônicas da nanoargila caulinita dopada com metal de transição
Resumo
Neste trabalho, uma série de nanoclays de caulinita dopados com metais de transição (Cr, Mn, Fe e Co) foram investigados por meio de cálculos da teoria do funcional da densidade (DFT). A influência da dopagem metálica na estrutura geométrica e na estrutura eletrônica da caulinita foi analisada. Foram estudados os estados ferromagnético (FM), antiferromagnético (AFM) e não magnético (NM) de estruturas de caulinita dopadas com metal de transição (TM). O volume do cristal, os parâmetros de rede, o comprimento da ligação, a carga e o spin foram calculados pela teoria do funcional da densidade corrigida por dispersão (DFT-D2). Os resultados indicaram que Cr 3+ e Fe 3+ dopantes mostraram-se mais estáveis sob o estado AFM, enquanto Mn 3+ preferiu os estados AFM e FM, e Co 3+ dopante estado NM preferido. Além disso, a dopagem do metal de transição pode induzir a expansão do volume da rede e alguns estados dopantes no gap.
Histórico
Minerais de nanoargila do grupo caulim, como resultado de alteração hidrotérmica e / ou processos de intemperismo, têm propriedades físicas únicas por causa de sua estrutura em camadas, tamanho de grão pequeno e, mais importante, superfície hidratada com muitos grupos hidroxila. Tem atraído a atenção de pesquisadores em química de materiais, química ambiental e física mineral [1,2,3,4,5,6,7,8,9,10,11]. A caulinita, um dos minerais de nanoargila mais abundantes na Terra, tem sido amplamente utilizada em plásticos, catálise e na indústria de cimento. A funcionalização adicional da caulinita como novos materiais de suporte tem atraído cada vez mais as atenções em vários campos. A caulinita pode simplesmente servir como materiais de suporte para se misturar com outras nanopartículas para formar materiais de mudança de fase para utilidade de energia solar [4, 5] ou revestidos com óxido dopado para formar pós condutores para aplicações em campos condutores [9, 12]. A hibridização de caulinita com nanopartículas funcionais foi encontrada para aumentar a atividade fotocatalítica de Pd-ZnO e as propriedades de luminescência de CdS através de um efeito sinérgico [6, 7]. As propriedades da superfície da caulinita foram modificadas pela ancoragem de alguns grupos funcionais na superfície [13, 14] ou por pré-tratamento de ativação com ácido para melhorias adicionais [2].
As estruturas e energética dos minerais do grupo caulim foram extensivamente investigadas experimentalmente [15,16,17] e teoricamente [18,19,20,21,22]. O estudo teórico da adsorção de metais pesados na superfície da caulinita foi estudado para adsorção de Cd, Cu, Hg e Ni (II) [23], em que a capacidade de adsorção da argila de caulinita para íons foi encontrada na ordem de Ni> Cu> Cd> Hg (II). A adsorção e difusão de Pb (II) [24, 25] e uranil [26] na superfície da caulinita (001) foram estudadas [24,25,26], e o comportamento de adsorção em sistema aquoso também foi relatado posteriormente [27, 28]. A influência da dopagem com Mg, Ca e Fe na superfície da caulinita e a subsequente adsorção e penetração de H 2 O no intercalar foram estudados [29]. As energias de adsorção de H 2 O em caulinitas dopados (001) foram encontrados menos do que em superfície não dopada. A estrutura eletrônica da caulinita com e sem defeitos intrínsecos foi estudada pelos funcionais da teoria funcional da densidade padrão (DFT) e funcionais híbridos [30]. No entanto, não até recentemente as evoluções da estrutura durante o processo de desidroxilação, desaluminação e condensação de sílica da caulinita foram modeladas por cálculos DFT [1, 31, 32]. A remoção de Al em materiais do grupo caulim alterou muito a geometria e as propriedades eletrônicas desses materiais de camada e melhorou seu efeito de suporte [1, 2].
A dopagem de metais, como um método bem conhecido para modificar a estrutura e propriedades de compostos, foi teoricamente estudada para Al 2 O 3 [33], TiO 2 [34], MOF [35] e outros sólidos [36]. Explorar as mudanças na estrutura e nas propriedades da nanoargila de caulinita sobre a dopagem com metal de transição (TM) seria interessante para este material argiloso em camadas. Neste trabalho, uma série de nanoargila de caulinita dopada com Cr, Mn, Fe e Co foi estudada por cálculos DFT e teve como foco a influência da dopagem metálica na estrutura geométrica e na estrutura eletrônica da nanoargila de caulinita. Os possíveis estados ferromagnético (FM), antiferromagnético (AFM) e não magnético (NM) dessas estruturas de caulinita dopada com metal de transição foram estudados. Os parâmetros de rede, comprimento de ligação, carga e spin foram otimizados e calculados pela teoria do funcional de densidade corrigida por dispersão (DFT-D2).
Métodos
Todos os cálculos foram realizados com o código do programa CASTEP (Cambridge Sequential Total Energy Package) [37], baseado no primeiro princípio DFT. A aproximação de gradiente generalizado (GGA) com o potencial de correlação de troca de Perdew, Burke e Ernzerhof (PBE) foi usada para os cálculos [38]. As correções de dispersão DFT-D2 de Grimme foram incluídas para contabilizar as interações de dispersão de Van der Waals [39]. Um corte de energia de 500 eV foi aplicado usando o formalismo de onda plana de pseudo-potencial ultrasoft [40]. A grade Monkhorst – Pack [41] com 2 × 2 × 3 k A malha de pontos foi usada para relaxação geométrica e cálculos de estrutura eletrônica. A energia total autoconsistente no estado fundamental foi efetivamente obtida pelo esquema de mistura de densidade [42]. Para as otimizações de geometria, o limite de convergência para tolerância de campo autoconsistente (SCF) foi definido como 1,0 × 10 −6 eV / átomo, todas as forças sobre os átomos convergiram para menos de 0,03 eV / Å, o tensor de tensão total foi reduzido para a ordem de 0,05 GPa e o deslocamento iônico máximo estava dentro de 0,001 Å. Os elementos investigados nos estados de valência foram O (2s 2 2p 4 ), Al (3s 2 3p 1 ), Cr (3s 2 3p 6 3d 5 4s 1 ), Mn (3d 5 4s 2 ), Fe (3d 6 4s 2 ) e Co (3d 7 4s 2 ) Os pseudo-potenciais do Uspcc foram usados para Mn, Fe e Co, e os pseudo-potenciais do usp para o resto dos elementos. Os parâmetros da célula e a coordenação atômica foram totalmente relaxados durante a otimização da geometria usando um algoritmo de minimização Broyden – Fletcher – Goldfarb – Shanno (BFGS). A simetria do cristal foi removida pela imposição de diferentes momentos magnéticos iniciais sobre os íons TM para que o estado fundamental eletrônico pudesse adotar uma simetria mais baixa.
Resultados e discussão
A estrutura inicial da caulinita foi obtida em nosso trabalho anterior [1]. A Figura 1 mostra a estrutura cristalina relaxada 2 × 2 × 1 da caulinita (4 unidades de caulinita). A estrutura da camada de caulinita, Al 2 Si 2 O 5 (OH) 4 , é composto por uma folha octaédrica de Al – O e uma folha tetraédrica de Si – O, conectada por um átomo apical de O (O a ) O tetraedro Si – O é construído por um átomo central de Si e quatro átomos circundantes de O, em que um é o O a átomo e os outros três são os átomos de O basais (O b ) O octaedro Al-O é construído por um Al central e seis O circundantes, em que dois são O a átomo e os outros quatro são átomos de O (em grupos OH) compartilhados com outro octaedro Al – O. Além disso, esses grupos OH podem ser divididos em dois tipos:o OH inter-camadas (OH inter ) na superfície da estrutura da camada e do OH interno (OH interno ) dentro da estrutura de camada entre a folha de Al e a folha de Si. Portanto, existem dois tipos de ligações Si – O, Si – O a e Si – O b (linha de ponto preto) e três tipos de ligações Al – O, Al – O inter (linha de ponto vermelho), Al – O interno (linha pontilhada verde) e Al – O (linha pontilhada preta) na estrutura volumétrica de caulinita.
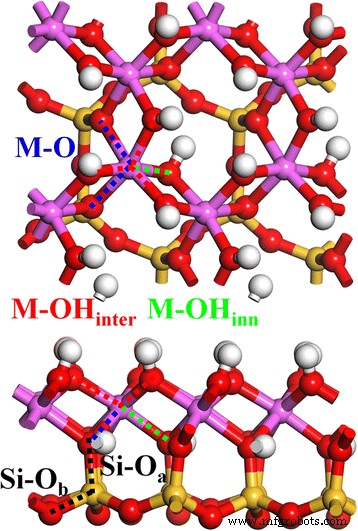
Superior ( para cima ) e lateral ( para baixo ) visualizações de caulinita. O Si – O a ( preto ), Si – O b ( preto ), M – OH inter ( vermelho ), M – OH interno ( verde ), e M – O ( azul ) obrigações são indicadas por linhas pontilhadas
A energia de dispersão sempre desempenha um papel importante na estabilização da estrutura do mineral de argila devido à interação entre as camadas [21, 43]. Entre os vários funcionais híbridos, PBE-D2 [21], B3LYP [22], B3LYP-D [18] e RPBE-D2 [18, 21], que foi usado para obter a estrutura de rede experimental da caulinita [44, 45 ], PBE-D2 funcional foi considerado preciso e menos demorado. A superestimativa do funcional PBE para comprimentos de ligação são superados pela correção de dispersão em comparação com os resultados experimentais, como brevemente relatado anteriormente [1]. A fim de distinguir o efeito do doping TM na estrutura da caulinita, aqui, primeiro revisitamos a estrutura da rede e as distâncias de ligação otimizadas entre os cátions centrais (Si e Al) e os átomos de oxigênio, O a , O b , e OH inn .
Conforme mostrado na Tabela 1, para caulinita, o volume de célula unitária calculado otimizado usando PBE-D2 com dispersão corrigida funcional está perto do valor experimental, o que dá erro relativo significativamente reduzido (∼0,4%) em comparação com PBE funcional (∼3,4%) . Para os vetores de rede aeb, o erro relativo usando PBE-D2 (∼0,4%) é muito menor do que PBE (∼1,1%). E, sob correções de dispersão de PBE-D2, a distância da camada (vetor c) de caulinita é diminuída em 0,17 Å (∼2%). Notavelmente, os ângulos de rede após a correção da dispersão estão muito próximos dos resultados experimentais, especialmente para α. Quanto às distribuições de comprimento de ligação em caulinita, embora PBE-D2 dê poucas melhorias para Si – O a , Al – OH interno , e ligações Al – O em comparação com os resultados experimentais, uma grande melhoria é feita para Al – OH inter ligação na superfície de Al – O (que é importante para a química de superfície) e ligeira melhora para Si – O b ligação na superfície de Si – O. Notavelmente, para Al – OH inter ligação, a correção de dispersão de PBE-D2 parece descrever com precisão o ambiente de ligação na camada mais externa da superfície de Al – O, que é fortemente influenciada pela força de dispersão da superfície de Si – O de outra camada de caulinita que fica acima. Outro ponto a ser mencionado aqui é que na verdade existem duas ligações divididas de Al – O (Fig. 1, linha pontilhada azul) com comprimentos de ligação significativamente diferentes de cerca de 1,95 e 2,00 Å [45], o que mostra a distorção da rede de Al – O octaedro originado da incompatibilidade de rede entre a folha de Si – O e a folha de Al – O. Como um grande erro no cálculo da estrutura da caulinita em comparação com os resultados experimentais, essas ligações Al-O são superestimadas por PBE e PBE-D2, com comprimento médio de ligação semelhante (Tabela 1). PBE-D2 dá duas ligações Al-O de aproximadamente 1,96 e 2,04 Å, com a segunda superestimada em 0,04 Å (Fig. 2, linha pontilhada azul).
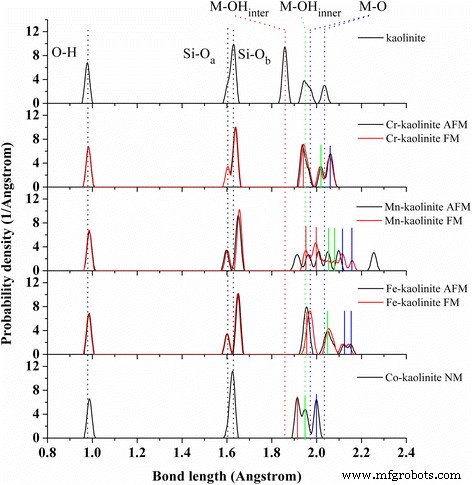
A distribuição de ligações de Cr–, Mn–, Fe– e Co – caulinita. Os estados multimagnéticos são dados para cada TM-caulinita. Os diferentes tipos médios de O – H ( preto ), Si – O a ( preto ), Si – O b ( preto ), M – OH inter ( vermelho ), M – OH interno ( verde ), e M – O ( azul ) as ligações na caulinita são indicadas por linhas pontilhadas . O M – OH inter ( vermelho ), M – OH interno ( verde ), e M – O ( azul ) ligações em Cr-caulinita ( AFM ), Mn-caulinita ( FM ), Fe-caulinita ( AFM ) e Co-caulinita ( NM ) são indicados por linhas sólidas
Os metais de transição (Cr, Mn, Fe e Co) caulinitas dopados foram construídos substituindo o átomo de Al pelo átomo de Cr, Mn, Fe ou Co. Apenas a substituição equivalente de Al 3+ íon com TM 3+ O íon foi considerado uma vez que a substituição não equivalente de íons TM com um estado químico diferente de +3 causará espaços vazios ou impurezas adicionais para o equilíbrio de carga. Do ponto de vista da estrutura, os funcionais PBE e PBE-D2 da TM-caulinita apresentam diferenças estruturais semelhantes às observadas para a caulinita. Considerando que o funcional PBE-D2 descreve melhor os vetores de rede e os comprimentos de ligação das duas superfícies basais da caulinita, após a discussão sobre a caulinita TM, dependeu principalmente dos resultados obtidos pelo funcional PBE-D2. Os parâmetros de rede, comprimento de ligação, carga e spin da caulinita dopada com TM e seus estados magnéticos foram resumidos na Tabela 1. As diferenças de energia (por átomo de TM) entre os estados de AFM e FM para Cr-caulinita, Mn-caulinita e Fe- caulinita são 0,022, -0,006 e 0,094 eV, respectivamente. Uma vez que a estrutura da Co-caulinita é estável apenas em um estado não magnético, apenas a estrutura NM da Co-caulinita é mostrada.
Os volumes de células unitárias de TM-caulinita são expandidos em comparação com a caulinita, com uma tendência de Mn-caulinita> Fe-caulinita>> Cr-caulinita>> caulinita> Co-caulinita. As expansões celulares são causadas principalmente pelas ligações M – O mais longas em comparação com as ligações Al – O, levando à maior expansão nos vetores de rede a e b. Enquanto isso, o Si – O b ligações na folha de Si – O são alongadas simultaneamente, e os ângulos de rede cristalina de α e β são distorcidos de acordo. O volume da célula de Mn-caulinita com o estado FM é aumentado em 1,4% em comparação com o estado AFM, enquanto em contraste, pouca influência da ordenação magnética nos volumes das células é encontrada para Cr-caulinita e Fe-caulinita. Os momentos magnéticos de Cr, Mn, Fe e Co são próximos aos do Al TM dopado 2 O 3 [33], enquanto a carga Mulliken é ligeiramente mais alta, o que implica uma reatividade mais forte.
As distribuições do comprimento da ligação da caulinita TM são analisadas na Fig. 2, com diferentes tipos de ligações Si-O e M-O na caulinita TM indicadas por linhas contínuas para cada elemento de dopagem. De modo geral, há um aumento dos comprimentos de ligação de M – O e Si – O b após o doping TM, e entretanto, há uma reorganização da distribuição das ligações das ligações M – O divididas para M – OH inter (vermelho), M – OH interno (verde) e ligações M – O (azul). Notavelmente, as ligações Al-O divididas (linha pontilhada azul) desapareceram após o doping com Cr e Co. Além disso, as distribuições de comprimento de ligação são altamente dependentes da ordem magnética para átomos de Mn, mas são apenas ligeiramente influenciadas para átomos de Cr e Fe.
Os resultados do PDOS para Cr 3+ (d3), Mn 3+ (d4), Fe 3+ (d5), e Co 3+ (d6) e as distribuições de densidade de carga correspondentes são mostradas nas Figs. 3 e 4. De acordo com o teorema de Jahn-Teller, qualquer sistema eletrônico degenerado irá distorcer espontaneamente de forma a remover a degenerescência [46], que é afetada pelo ambiente de ligação circundante [47]. Para TM 3+ dopagem no sítio Al octaédrico de caulinita com muitos grupos hidroxila, os cinco orbitais d-shell do TM 3+ irá se dividir em um trio t 2g estado e um dupleto e g estado em Oh simetria. Os elétrons no estado tripleto estão localizados na região intermediária entre os ligantes e posteriormente hibridizados com os estados O mais próximos. Aqueles no estado de dupleto apontam diretamente para os ligantes e, portanto, têm mais energia do que t 2g elétrons. Geralmente, a presença de elétrons no e g orbitais tendem a desestabilizar a ligação octaédrica, e a degeneração é removida alongando as ligações opostas ao orbital preenchido e encurtando as ligações opostas ao orbital vazio. A transição d – d de TM 3+ (Oh) espécie é sempre do t 2g ocupado orbitais (dxy, dyz e dzx) para e g desocupado orbital (d x2-y2 ou d z2 , dependendo de sua ocupação). A divisão orbital entre e g orbitais e t 2g orbitais de Cr 3+ (d 3 ), Mn 3+ (d 4 ), Fe 3+ (d 5 ), e Co 3+ (d 6 ) na TM-caulinita é semelhante àquela em Al 2 O 3 e TiO 2 [33, 48, 49], mas as energias de divisão entre os orbitais 3d são ligeiramente maiores do que em seus próprios óxidos (Fig. 3), possivelmente devido à hibridização com os grupos hidroxila circundantes.
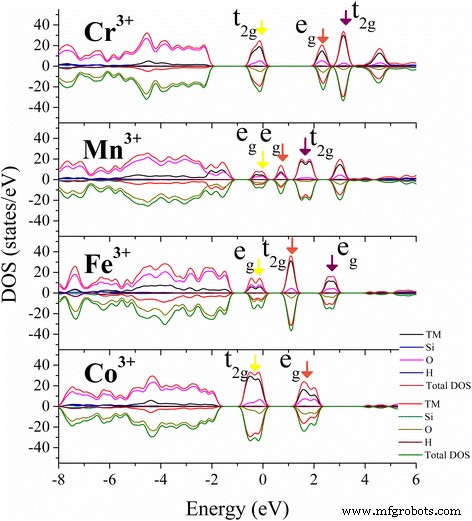
São fornecidas a densidade total de estados (DOS) e a densidade de estados projetados em átomos (PDOS) dos estados mais estáveis para caulinita dopada com TM. Os orbitais 3d mais ocupados ( amarelo ) e o primeiro ( marrom ) e segundo ( roxo ) os orbitais 3d mais baixos desocupados em torno do nível de Fermi são apontados por setas coloridas
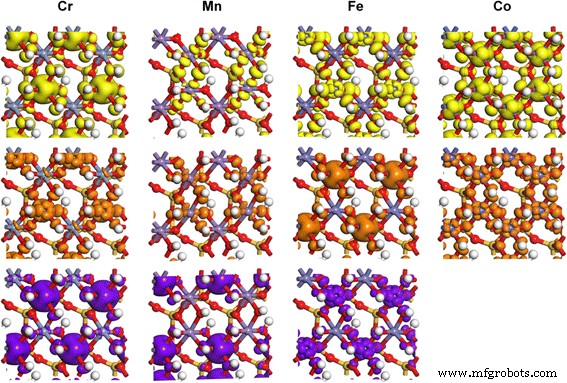
Densidade de carga parcial de orbitais TM 3d em TM-caulinita, correspondendo aos estados apontados por setas nos resultados do PDOS. Os níveis de isosuperfície são 0,02 e / Å 3
A diferença de divisão de energias entre os estados FM e AFM de Mn-caulinita é pequena e as distribuições de densidade de estados são semelhantes, exceto as direções de spin são diferentes. Portanto, para simplificar, apenas os resultados para o estado AFM são mostrados. Para o Mn de alta rotação 3+ (d 4 ) íon em Mn-caulinita com estado AFM, apenas um dos dois e g orbitais é ocupado no máximo da banda de valência (VBM) (Fig. 3, seta amarela). A ocupação de d z2 orbital que é mais baixo em energia dá uma forte repulsão nos elétrons de ligação dos dois ligantes ao longo do z eixo e alonga a ligação M – O nessa direção. Este efeito é o conhecido efeito Jahn-Teller. Os estados na parte inferior do mínimo da banda de condução (CBM) são compostos pelo mais baixo desocupado d z2 orbital (seta marrom) e o d x2y2 superior orbital (seta roxa) de Mn 3+ (d 4 ) Para Cr 3+ (d 3 ), Fe 3+ (d 5 ), e Co 3+ (d 6 ) caso dopado, onde o t 2g e e g orbitais são ocupados uniformemente, a influência do efeito de distorção de Jahn-Teller é pequena, o que causou apenas um ligeiro desvio das ligações M-O na TM-caulinita (Fig. 2). Tal modificação de estrutura e propriedades eletrônicas por dopagem de TM pode melhorar a aplicação de caulim no campo da catálise [50, 51], captura de CO [52, 53], carga de droga [54] e armazenamento de energia [55,56,57 ] E, também pode ser aplicado a outros minerais, como montmorilonita [50, 58], perlita [55] e talco [59] para alterar suas propriedades eletrônicas.
Conclusões
A influência da dopagem de metais de transição (Cr, Mn, Fe e Co) na estrutura geométrica e na estrutura eletrônica da nanoargila de caulinita é investigada por cálculos DFT. O volume do cristal, os parâmetros da rede, o comprimento da ligação, a carga e o spin e os possíveis estados magnéticos são calculados e estudados. O Cr 3+ e Fe 3+ os dopantes mostram-se mais estáveis no estado AFM, Mn 3+ prefira o estado FM e Co 3+ os dopantes preferem o estado NM. A dopagem do metal de transição induz a expansão do volume da rede e alguma reorganização das distribuições de ligações M – O. Enquanto isso, os dopantes TM introduzem alguns estados 3d com energias de divisão maiores no gap de caulinita.
Abreviações
- AFM:
-
Antiferromagnético
- BFGS:
-
Broyden – Fletcher – Goldfarb – Shanno
- CASTEP:
-
Pacote de energia total sequencial de Cambridge
- CBM:
-
Banda de condução mínima
- DFT:
-
Teoria da densidade funcional
- DFT-D2:
-
Teoria do funcional da densidade corrigida por dispersão
- FM:
-
Ferromagnético
- GGA:
-
Aproximação de gradiente generalizado
- NM:
-
Não magnético
- PBE:
-
Perdew, Burke e Ernzerhof
- SCF:
-
Campo autoconsistente
- TM:
-
Metal de transição
- VBM:
-
Banda de valência máxima
NiCo2S4 @ NiMoO4 Core-Shell Heterostructure Nanotube Arrays cultivados em Ni Foam como um eletrodo livre de aglutinante exibiu alto desempenho eletroquímico com alta capacidade
Um breve relatório de progresso sobre células solares de perovskita de alta eficiência
Nanomateriais
- Revelando a estrutura atômica e eletrônica das nanofibras de carbono de copo empilhado
- Modulação das propriedades de anisotropia eletrônica e óptica de ML-GaS por campo elétrico vertical
- Estados eletrônicos do nanocristal dopado com oxigênio e emissão visível em silício negro preparado por ns-Laser
- Influência da Água na Estrutura e Propriedades Dielétricas da Microcristalina e Nano-Celulose
- Propriedades ópticas e eletrônicas de fotodiodos N + / P de silício hiperdopado com enxofre induzido por laser de femtosegundo
- Sondando as propriedades estruturais, eletrônicas e magnéticas dos aglomerados Ag n V (n =1–12)
- Morfologia, estrutura e propriedades ópticas de filmes semicondutores com Nanislands GeSiSn e camadas deformadas
- 20 tipos diferentes de metal e suas propriedades
- Metal de cromo:elementos, propriedades e usos
- Propriedades de PCB automotivo e considerações de design