


Identificação de uma via de dissociação de excitons intramolecular específica da ponte em polímeros conjugados alternados doador-π-aceitador
Resumo
A dissociação intramolecular de excitons é crítica para gerações de portadores de carga móvel altamente eficientes em células solares orgânicas. No entanto, apesar de muita atenção, os efeitos das pontes π na dinâmica de dissociação de excitons em polímeros conjugados alternados doador-π-aceitador (D-π-A) permanecem ainda obscuros. Aqui, usando uma combinação de espectroscopia de absorção transiente (TA) resolvida no tempo de femtossegundo e espectroscopia de estado estacionário, rastreamos a dinâmica de relaxamento de exciton intramolecular ultra-rápida em três polímeros conjugados alternados D-π-A que foram sintetizados pelo grupo de Qin e denominados HSD-A , HSD-B, HSD-C. Verificou-se que a adição da unidade de tiofeno como pontes π levará ao desvio para o vermelho do espectro de absorção em estado estacionário. É importante ressaltar que revelamos a existência de uma nova via de dissociação do exciton intramolecular mediada por um estado de transferência de carga específica de ponte (CT ′) com o pico da impressão digital TA em 1200 nm em HSD-B e HSD-C com ponte π. Este estado CT ′ resulta em taxas de captura de elétrons mais altas para HSD-B e HSD-C em comparação com HSD-A. Dependendo da proporção do estado CT ′ e a recombinação não-geminada são etapas importantes para a compreensão das eficiências de conversão de alta potência em HSD-B do que em HSD-C. Propomos que esta via de dissociação de excitons específicos da ponte desempenha um papel importante na dissociação ultra-rápida de excitons intramoleculares de polímeros conjugados alternados de material fotovoltaico orgânico D-π-A.
Introdução
Dispositivos fotovoltaicos orgânicos (OPV) que usam energia solar para atender à crescente demanda mundial de energia podem ser considerados um dos mais importantes substitutos para a produção de fontes de energia limpa e renovável [1,2,3,4]. Polímeros conjugados alternados doador-aceitador, nos quais blocos conjugados de afinidade eletrônica diferente são arranjados alternadamente ao longo da cadeia principal dos polímeros, são excelentes materiais eletrônicos orgânicos. O fato de que ele mostra uma eficiência de conversão de energia relativamente alta (PCE) é em parte devido às lacunas ópticas mais baixas que permitem uma coleta mais eficiente de fótons solares na faixa do infravermelho próximo (NIR) [5]. Em conseqüência, os dispositivos OPV que consistem em polímeros conjugados alternados doador-aceitador podem se tornar as alternativas economicamente viáveis para células solares baseadas em Si [6,7,8,9].
As pesquisas mais recentes de vários grupos mostraram que a eficiência da OPV pode ser melhorada usando copolímeros alternados conjugados com gap de banda baixa (como visto nas famílias PCDTBT, PBDTTT e PTB) [10,11,12,13]. Uma diferença importante entre esses polímeros é que a transição do exciton no nível de energia mais baixo exibe características parciais de transferência de carga. O estado de transferência de carga intramolecular é considerado promotor da separação final de carga na heterojunção [14,15,16,17,18,19]. Portanto, é razoável esperar que as propriedades dos polímeros doadores de baixo bandgap estejam intimamente relacionadas ao desempenho dos dispositivos. No entanto, a conexão entre o desempenho dos dispositivos e as propriedades inerentes dos polímeros ainda é indistinta. Especialmente, a divisão ultrarrápida do exciton e a dinâmica da portadora de polímeros doadores de baixo bandgap não estão diretamente relacionadas ao PCE dos dispositivos. Por um lado, uma massa de curso paralelo e sequencial em escala temporal ultrarrápida e lenta só é encontrada em condições relacionadas ao equipamento. Por outro lado, apenas a divisão do exciton na interface doador-aceitador da heterojunção em massa (BHJ) poderia ser considerada para condicionar o dispositivo PCE [5]. Portanto, é necessário estudar a dinâmica do exciton e da portadora de polímeros doadores de baixo bandgap para otimizar o PCE de dispositivos fotovoltaicos orgânicos D-A.
Uma série de polímeros conjugados alternados D-π-A foram recentemente sintetizados pelo grupo de Qin [20,21,22]. Por exemplo, os copolímeros HSD consistem em carbazol ligado a 2,7 como unidade doadora e 5,6-bis (octiloxi) benzo [c] [1,2,5] tiadiazol como unidade aceitadora, embora tenham diferentes pontes π. O bloco doador e o bloco aceitador são polimerizados diretamente para preparar o material fotovoltaico como HSD-A. Diferentemente, uma unidade de tiofeno atua como o bloco dador de conexão da ponte π e o bloco aceitador é denotado como HSD-B, bem como o bloco doador e o bloco aceitador são conectados por duas unidades de tiofeno é denotado como HSD-C. Eles descobriram que as pontes π nos copolímeros têm um efeito significativo nas propriedades dos copolímeros HSD. Diferentes pontes π afetam criticamente a deslocalização de elétrons da cadeia principal do polímero conjugado, a morfologia do filme e as propriedades ópticas, eletroquímicas, de transporte de carga e fotovoltaicas dos copolímeros HSD [23]. Usando copolímeros HSD como doadores de elétrons e PC 71 BM como aceptor de elétrons para preparar dispositivos fotovoltaicos orgânicos, verifica-se que os dispositivos preparados com polímeros HSD com diferentes pontes π possuem diferentes PCE. Dispositivos OPV com HSD-A:PC71BM como a camada ativa demonstrou que o PCE é baixo; HSD-B:PC71BM exibe um PCE de 5,4%; HSD-C:PC71BM mostra um PCE de 2,15% [20, 21]. Essas evidências indicam que os copolímeros doadores têm efeito sobre o PCE de células solares poliméricas, mas a correlação entre o desempenho dos dispositivos e as características inerentes aos copolímeros doadores como estrutura, dinâmica energética e de portadores móveis ainda é indistinta. Os principais processos de relaxamento que seguem a absorção de luz são importantes para determinar o desempenho dos dispositivos fotovoltaicos. Portanto, é imperativo compreender a dinâmica do exciton e da portadora de copolímeros de HSD rastreando excitons.
O rápido desenvolvimento da tecnologia de laser ultracurto torna possível monitorar e rastrear a formação e quebra de ligações químicas em moléculas e vários processos dinâmicos dentro e entre moléculas com resolução de tempo de femtossegundo e alta precisão espacial. Este trabalho elucida os processos de dissociação de excitons e relaxamento ultra-rápido de copolímeros de HSD usando uma combinação de absorção em estado estacionário e espectroscopia de absorção transiente resolvida no tempo de femtossegundo. As bandas de espectro características foram medidas e analisadas em detalhes, revelando um mecanismo de relaxamento ultrarrápido para a dinâmica de dissociação de excitons. Nossos resultados fornecem uma visão melhor sobre as propriedades físicas dos copolímeros HSD e fornecem uma base experimental para melhorar o PCE de células solares de polímero.
Materiais e métodos experimentais
Materiais
HSD-A, HSD-B, HSD-C foram fornecidos pelo grupo de Qin, e a síntese e caracterização desses coligômeros foram mostradas na literatura [20, 21]. As estruturas moleculares desses co-oligômeros são mostradas na Fig. 1a. A solução utilizada para a preparação desses co-oligômeros foi o-diclorobenzeno, com concentração de cerca de 0,1 mg / ml. Esta concentração pode não apenas garantir que um bom sinal resolvido no tempo possa ser medido, mas também pode garantir que o cromóforo esteja totalmente separado, de modo que o estado excitado não seja extinto sob a intensidade de excitação usada [24].
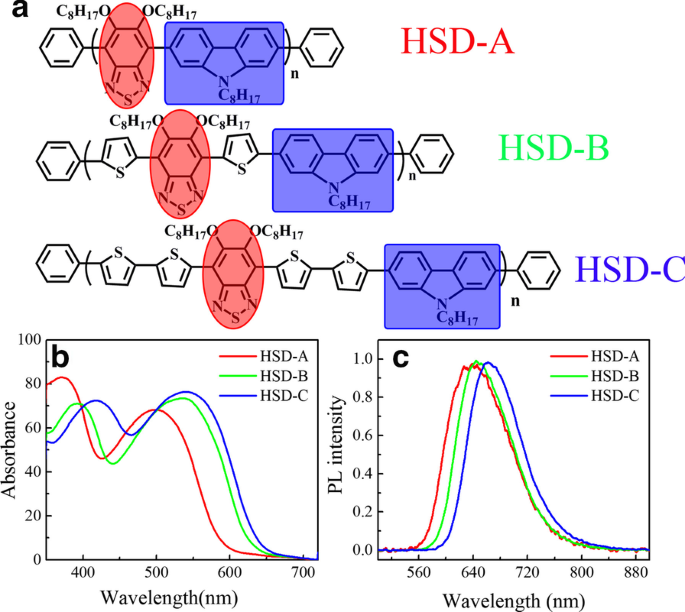
Estruturas moleculares de três polímeros ( a ) neste trabalho. Azul sombreado e vermelho sombreado denotam as partes doadora e aceitadora, respectivamente. Espectro de absorção em estado estacionário ( b ) e espectros de fotoluminescência em estado estacionário ( c ) de três amostras, HSD-A (vermelho), HSD-B (verde), HSD-C (azul), medido em o-diclorobenzeno
Medições espectroscópicas
A espectroscopia de absorção em estado estacionário foi medida por um espectrofotômetro de feixe duplo (Cary-5000, Agilent), e a espectroscopia de fluorescência em estado estacionário foi medida por um espectrômetro de fibra óptica (USB-4000, Ocean Optics).
A espectroscopia de absorção transiente resolvida no tempo de femtossegundo foi medida por laser de femtossegundo (Coerente), amplificador paramétrico óptico (OPA, TOPAS) e espectrômetro de absorção transiente (fogo Helios). O laser de femtossegundo gerado pelo laser de femtossegundo é dividido em dois caminhos através de um divisor de feixe (1:1), um dos quais entra em TOPAS e gera pulsos de bomba com comprimentos de onda diferentes; o outro feixe passa por um divisor de feixe (2:98) novamente e a pequena parte do laser projetado entra no espectrômetro de absorção transiente Helios para gerar pulsos de sonda de luz branca contínua (WLC) (420-780 nm, 820-1600 nm )
Resultados e discussão
A Figura 1a mostra a fórmula estrutural dos polímeros conjugados de HSD usados neste trabalho, as frações doadoras são marcadas com uma caixa azul e as frações aceitadoras são destacadas com um círculo vermelho. A unidade de tiofeno atua como a ponte específica entre o doador e o aceitador, evitando sequencialmente a repulsão estérica entre o doador individual e as unidades aceitadoras. Como relatado anteriormente, ele também pode alcançar a separação de carga de longa distância entre o doador e o aceitador, garantindo assim um estado de transferência de carga de longa duração [8]. A Figura 1b exibe os espectros de absorção em estado estacionário dos três polímeros, e os espectros de absorção dos três polímeros com diferentes pontes π apresentam formas semelhantes, caracterizadas por duas bandas de absorção distintas. O perfil típico de dois picos também foi relatado em outros polímeros conjugados, que são características únicas para polímeros conjugados D-A [25,26,27,28,29,30]. Os picos de absorção do HSD-A estão em torno de 370 e 490 nm, e os de HSD-B estão em torno de 390 e 530 nm, e os de HSD-C estão em torno de 420 e 540 nm. Esses dois picos de absorção são atribuídos à transição π – π * com o pico de energia inferior associado à transferência de carga intracadeia [31]. As posições dos picos de absorção em estado estacionário são afetadas pela substituição de diferentes unidades de pontes π, resultando no redshift dos picos de absorção principalmente por conta do efeito de deslocalização de elétrons [32]. Realizamos cálculos de química quântica em polímeros, e os orbitais moleculares de fronteira de polímeros HSD foram calculados e fornecidos na Fig. 2a. O orbital molecular mais ocupado (HOMO) e o orbital molecular mais baixo não ocupado (LUMO) das três amostras são mostrados na Fig. 2a, e as lacunas de energia HOMO – LUMO (ΔE H – L ) são plotados na Fig. 2b. Pode ser visto claramente na Fig. 2b que os hiatos de energia HOMO – LUMO (ΔE H – L ) de HSD-A para HSD-B e HSD-C diminuem gradualmente, o que é consistente com o desvio para o vermelho nos espectros de HSD-A para HSD-B e HSD-C [33]. A Figura 1c apresenta os espectros de fotoluminescência (PL) das três amostras em sua solução. Os espectros de PL das três amostras são semelhantes e consistentes com os espectros de absorção em estado estacionário. É digno de nota que seus picos se movem em direção à onda longa com o aumento do número de tiofeno. As pontes π de polímeros HSD podem sintonizar o PCE de células solares orgânicas fabricadas pela mistura de polímero HSD e PC71BM, o PCE dos dispositivos que usamos neste estudo está listado na seguinte ordem:HSD-B> HSD-C> HSD- A [20, 21].
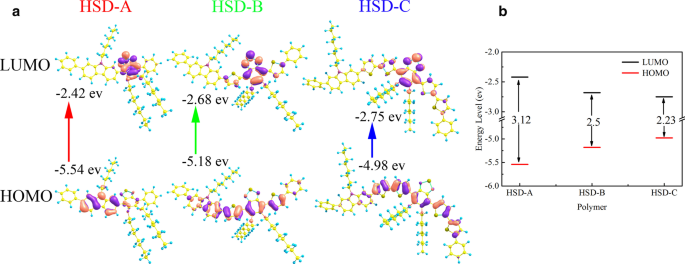
Orbitais moleculares de fronteira de copolímeros HSD ( a ) calculado usando o B3LYP-D3 funcional com conjunto de base 6-311G ** e os níveis de energia HOMO e LUMO ( b )
A espectroscopia de estado estacionário pode fornecer apenas uma macro-descrição dos estados de transição eletrônicos gerais. A fim de investigar como as pontes π afetam o PCE dos dispositivos, conduzimos ainda medições de absorção transiente dos três polímeros HSD, conforme mostrado na Fig. 3. Os gráficos das faixas do visível (VIS) das três amostras (Fig. 3a– c) são semelhantes, apresentando três características espectrais. O sinal negativo (azul claro no mapa) em cerca de 500 nm é atribuído ao sinal de branqueamento do estado fundamental (GSB), porque corresponde bem ao segundo pico de absorção constante, conforme mostrado na Fig. 1b. Todas as três amostras apresentam dois sinais de absorção positivos (vermelho claro no mapa) na faixa do visível, e os picos de absorção estão em 600 nm e 750 nm, respectivamente, considerados como absorção de estado excitado (ESA) [34]. Na faixa de detecção de infravermelho próximo (NIR) (Fig. 3d-f), as três amostras mostram diferenças óbvias. O HSD-A quase não tem sinal de absorção na faixa do infravermelho próximo, mas o HSD-B e o HSD-C têm sinal de absorção de grande área vermelho no escopo de 800–1500 nm.
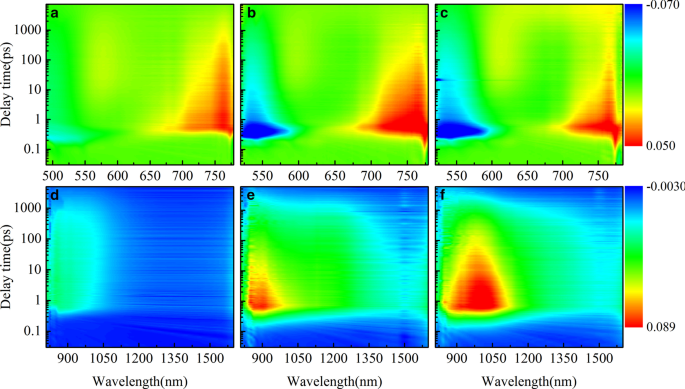
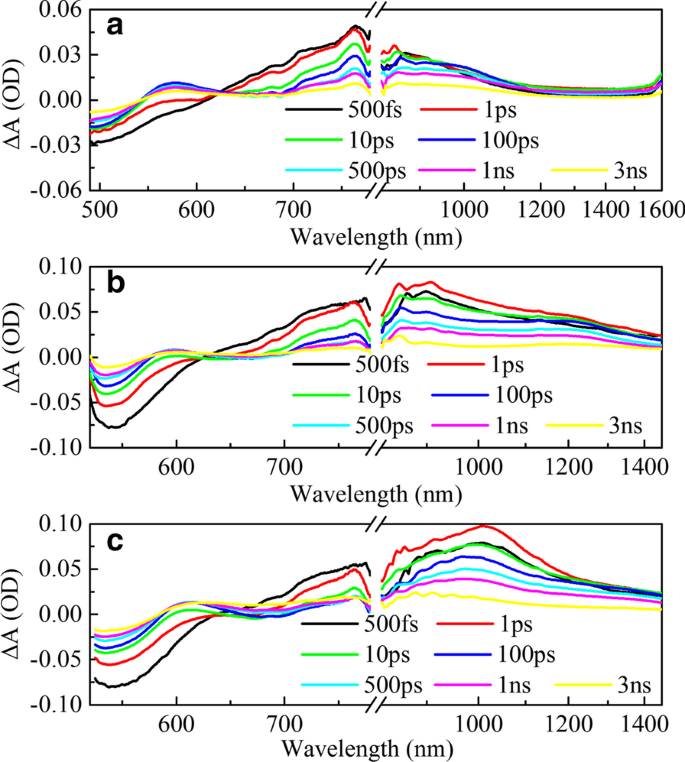
Espectros de absorção transiente (TA) resolvidos no tempo de femtossegundos em diferentes comprimentos de onda de sonda de diferentes amostras. Mapas bidimensionais como uma função do comprimento de onda da sonda (500-1600 nm) na excitação no comprimento de onda de 470 nm do HSD-A ( a , d ); excitação no comprimento de onda de 500 nm do HSD-B ( b , e ); excitação no comprimento de onda de 500 nm do HSD-C ( c , f )
A Figura 4 exibe a evolução temporal dos espectros de absorção diferencial dos três polímeros HSD em escala de tempo ultrarrápida. Na faixa de 490-780 nm, a excitação com baixa energia de fótons induz um amplo sinal positivo na região espectral de 700-780 nm que aumenta prontamente com o sinal negativo na região espectral de 490-600 nm como GSB. Atribuímos o sinal positivo amplo de 700 a 780 nm para a absorção do estado de exciton (EX) é atribuída como segue. Em primeiro lugar, seu tempo de vida é consistente com o tempo de vida de outros excitons nos polímeros isolados na literatura de 500-1000 ps [35] e é muito mais curto do que o tempo de vida do estado de transferência de carga (CT) e do estado de separação de carga (CS) , que tem uma escala de tempo maior do que alguns nanossegundos. Em segundo lugar, tem uma tendência dinâmica semelhante ao GSB nas primeiras centenas de picossegundos. Outro sinal positivo (com pico em cerca de 600 nm) aparece após alguns picossegundos, o que corresponde à formação do estado de portadora móvel (MC). Uma vez que os espectros de cerca de 600 nm podem ser razoavelmente atribuídos ao fato de que a superposição da absorção de GSB e MC nos experimentos de absorção transiente. O sinal negativo inicial em 600 nm deve-se ao fato do sinal do GSB ser muito mais forte do que o sinal de absorção do MC. À medida que o tempo de atraso aumenta, o recurso TA positivo aparece quando a absorção do MC é mais forte do que a do GSB. Além disso, o motivo do afundamento em 650 nm é que o estado excitado instável retorna ao estado fundamental devido à emissão estimulada (SE) e à consistência espectral com a fluorescência em estado estacionário. Nas faixas de NIR, pode-se observar que os sinais de absorção das três amostras aumentaram em 1 ps com um pico em cerca de 1 ps e depois mostram uma tendência a serem atenuados. Curiosamente, a forma e a tendência de atenuação dos sinais de absorção das três amostras são diferentes. A fim de analisar essas diferenças em mais detalhes, realizamos o ajuste de pico no espectro infravermelho e os resultados são mostrados na Fig. 5.
Espectros de diferença associada à evolução (EADS) em VIS – NIR de HSD-A ( a ), HSD-B ( b ), HSD-C ( c )
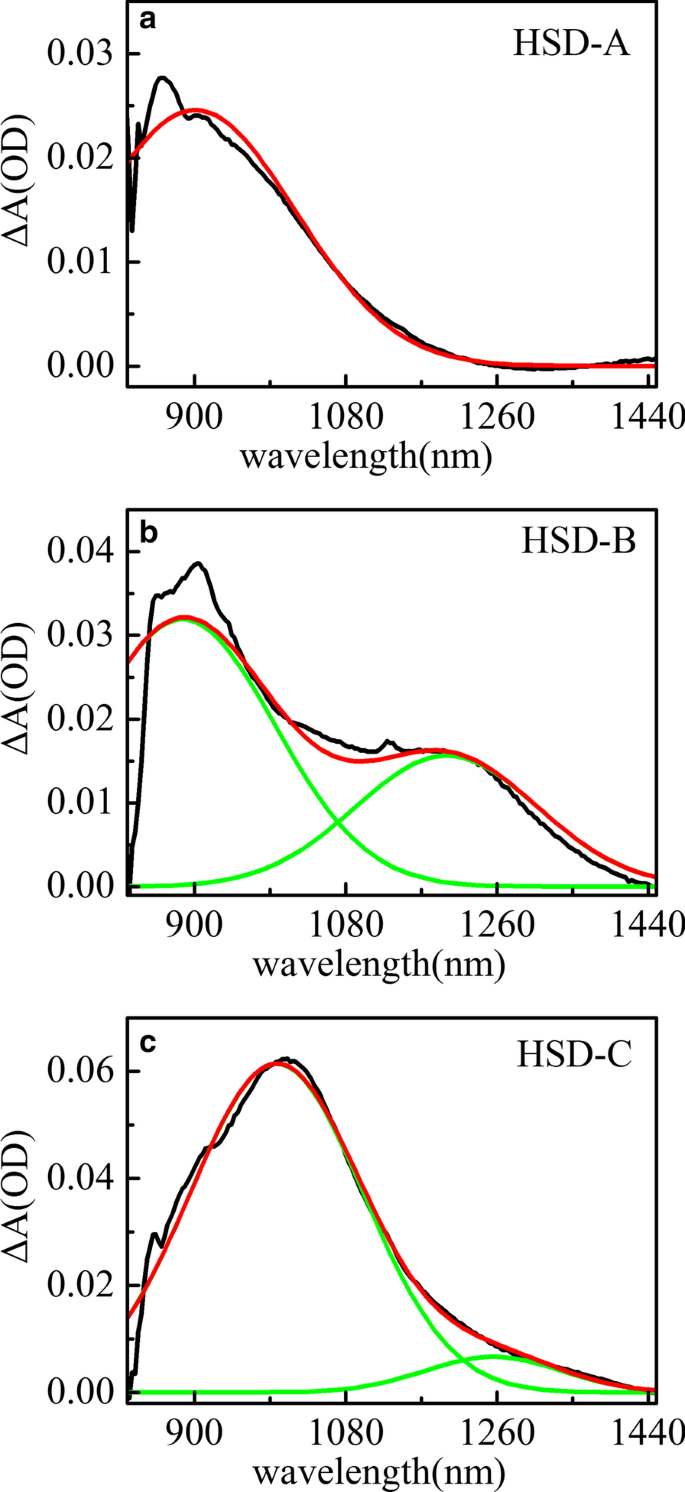
Pico espectral de absorção transiente ajustado de HSD-A ( a ) e HSD-B ( b ) e HSD-C ( c ) no tempo de atraso de 2 ps na faixa do infravermelho. A curva preta representa o espectro de absorção das amostras a 2 ps, a curva vermelha é o espectro de absorção ajustado e a verde é o sinal espectral identificado no espectro
A Figura 5 mostra o ajuste do pico do espectro de absorção transiente de HSD-A (a) e HSD-B (b) e HSD-C (c) no tempo de atraso de 2 ps na faixa do infravermelho. Em HSD-A, os espectros podem ser bem ajustados por uma análise de componente, enquanto em HSD-B e HSD-C, esses espectros podem ser mais aproximados por duas análises de componentes diferentes. Isso significa que HSD-B e HSD-C possuem um sinal a mais de absorção na faixa do infravermelho do que a dose de HSD-A, que se deve à adição da ponte de tiofeno. A adição da ponte de tiofeno expande a faixa de absorção dos polímeros em NIR, permitindo que HSD-B e HSD-C tenham um novo pico de absorção próximo a 1200 nm. No início, esses dois sinais positivos têm um tempo de subida intimamente relacionado à atenuação dos picos EX, conforme mostrado na Fig. 6, que mostra que esses sinais positivos são gerados diretamente pelos estados EX. Os sinais positivos próximos a 900 nm em todas as três amostras podem ser alocados ao estado de transferência de carga intramolecular (CT). Nesse estado, os excitons se dividem em pares de elétron-buraco, e os pares de elétron-buraco ainda estão próximos o suficiente para gerar a gravidade de Coulomb [36, 37]. Outros novos sinais positivos próximos a 1200 nm estão presentes apenas em HSD-B e HSD-C e também são acompanhados pela atenuação de EX no período inicial, mas têm uma tendência de atenuação diferente do estado de CT sob uma janela de tempo longa. Este é um novo canal de dissociação de excitons, e o consideramos como o estado CT ′. Porque tem características semelhantes ao estado do TC, mas a dinâmica de decaimento é diferente do estado do TC.
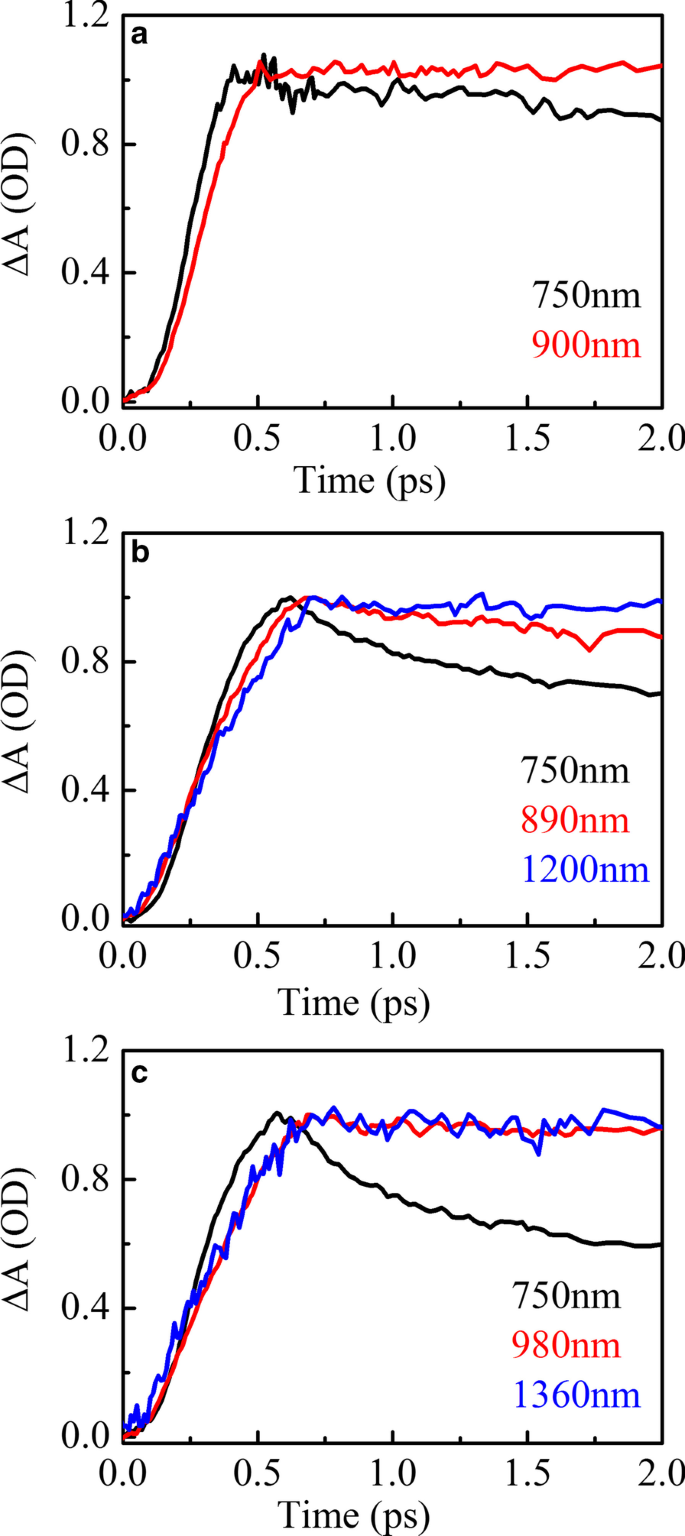
Absorção transiente EX (preto) e CT (vermelho) e CT ′ (azul) cinética para HSD-A ( a ) e HSD-B ( b ) e HSD-C ( c ), exibindo o decaimento EX simultaneamente com o aumento em CT e CT ′
Conforme mostrado na Fig. 7, as curvas dinâmicas do estado MC, estado EX, estado CT e CT ′ em HSD-A, HSD-B e HSD-C são extraídas e ajustadas, respectivamente, que representam a evolução do tempo de diferentes componentes. A fórmula de ajuste para as curvas dinâmicas é ∆A (t) =a 1 exp (- t / τ 1 ) + A 2 exp (- t / τ 2 ) + ··· + a n exp (- t / τ n ), onde um 1 , um 2 ,… A n são amplitudes, τ 1 , τ 2 ,…, Τ n correspondem às constantes de tempo [38, 39]. A Tabela 1 lista os componentes de tempo ajustados e amplitudes relativas. Para efeito de comparação, as amplitudes máximas são normalizadas. Pode-se ver claramente a partir dos dados de ajuste do estado MC (Fig. 7a, b) que a velocidade de geração de portadora do HSD-A é a mais rápida. Sua vida de formação é de 6,43 ps, que é menor que 12,6 ps de HSD-B e 8,41 ps de HSD-C. No entanto, os portadores de HSD-A decaem rapidamente após serem formados, enquanto HSD-B e HSD-C têm um processo de aumento lento adicional com a constante de tempo de 28,8 ps e 26,4 ps, respectivamente. Isso poderia tornar as portadoras em HSD-A mais difíceis de serem capturadas, o que provavelmente é uma das razões para o PCE mais baixo dos dispositivos. No estado EX (Fig. 7c, d), as tendências de decaimento das três amostras são obviamente diferentes. HSD-A tem uma vida de decaimento significativamente mais longa, então a divisão do exciton é relativamente lenta. Em HSD-B e HSD-C, há três tempos de vida de decaimento em 1 ns, um femtossegundo (0,712 ps para HSD-B, 0,408 ps para HSD-C) e um picossegundo (18,4 ps para HSD-B, 7,96 ps para HSD-C) os tempos de vida representam a transição do estado EX para outros estados. O tempo de vida mais longo de centenas de picossegundos (735 ps para HSD-B, 627 ps para HSD-C) é da mesma ordem de magnitude que o tempo de vida do exciton previamente relatado de P3HT isolado. Portanto, pode-se considerar que \ (\ uptau _ {3} ^ {{\ text {EX}}}} \) no ajuste EX tem mais probabilidade de ser o tempo de vida do exciton sem o processo de transição [35, 40] . No entanto, em uma janela de tempo longa, a recombinação de excitons ocorre em HSD-C. A cinética CT (Fig. 7e, f) dessas três amostras é melhor ajustada por três componentes de vida. A constante de tempo crescente curta, \ (\ uptau _ {1} ^ {{\ text {CT}}}} <1 \) ps, está intimamente relacionada ao tempo de vida de decaimento simultâneo do estado EX, o que significa a transição de Estado EX para estado CT. Da mesma forma, também há uma boa relevância entre o tempo de vida de decaimento do estado CT \ (\ uptau _ {2} ^ {{{\ text {CT}}}} \) e o aumento do tempo de vida do estado da portadora \ (\ uptau _ {2} ^ {{{\ text {MC}}}} \), indicando a transição do estado CT para o estado MC. Comparando os tempos de vida de decaimento do estado de CT das três amostras, pode-se descobrir que os tempos de vida de decaimento do estado de CT de HSD-B são significativamente mais curtos do que aqueles de HSD-A e HSD-C, indicando que HSD-B tem um decaimento de estado de CT mais rápido avaliar. O estado CT ′ (Fig. 7g, h) de HSD-B e HSD-C exibe curvas dinâmicas diferentes do estado CT no intervalo de 50 ps. Tem uma vida útil de transição mais longa \ (\ uptau _ {2} ^ {{{\ text {CT}} ^ {{\ prime}}}} \), que está em boa correlação com \ (\ uptau _ {3} ^ {{{\ text {MC}}}} \), que representa a transição do estado CT ′ para o estado MC.
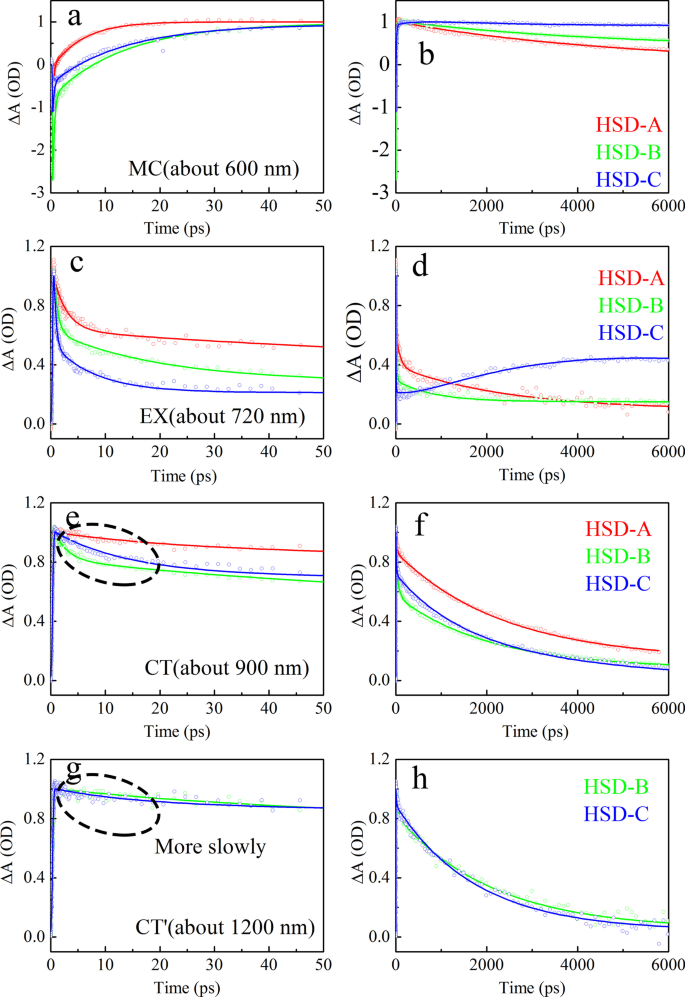
Ajuste cinético de todas as características espectrais transitórias de três amostras. A figura mostra a cinética de 50 ps ( a , c , e ) e 5000 ps ( b , d , f ) Os ajustes são para MC ( a , b ), EX ( c , d ), CT ( e , f ), CT ′ ( g , h ) características do espectro
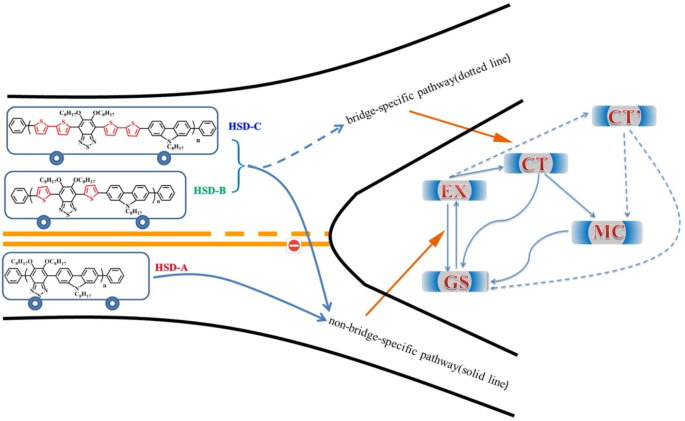
Um esquema do diagrama de energia simplificado para as vias de relaxamento de excitons é proposto na Fig. 8. A diferença na conformação local nos polímeros leva à variação de energia em diferentes estados. Portanto, esses estados apresentam características diferentes de TA. Em polímeros HSD, o estado MC, o estado EX, o estado CT e o estado CT ′ são inevitavelmente propostos para dar uma explicação mais razoável para o mecanismo de relaxamento do exciton. Excitons gerados após a excitação de luz irão rapidamente se dividir em estado CT e estado CT ′. Nesses estados, os excitons se dividem em pares buraco-elétron e ainda estão próximos o suficiente para sentir a atração coulombiana. Com o retardo de tempo, o buraco do elétron do estado CT e do estado CT ′ continuará a se dividir em estados MC mais estáveis. É importante notar que descobrimos que o estado CT ′ só existe em HSD-B e HSD-C com pontes de tiofeno, o que adiciona um novo canal de divisão de excitons aos polímeros HSD. Isso resultará em taxas de captura de elétrons mais altas para HSD-B e HSD-C, o que é consistente com o PCE mais alto de HSD-B e HSD-C em comparação com HSD-A. Enquanto isso, o fato de que o PCE de HSD-C é menor do que o de HSD-B poderia ser razoavelmente explicado da seguinte forma:(a) a adição de dois tiofenos como ponte π pode aumentar a separação espacial entre D e A, resultando em não geminação recombinação em HSD-C. (b) Como visto na Fig. 5, a proporção do estado CT ′ de HSD-B é significativamente maior do que a de HSD-C, o que determina todo o PCE de HSD-B. Portanto, o estado CT ′ em polímeros HSD é crítico para uma maior capacidade de captura de elétrons para resultar em um PCE mais alto de dispositivos de polímero HSD.
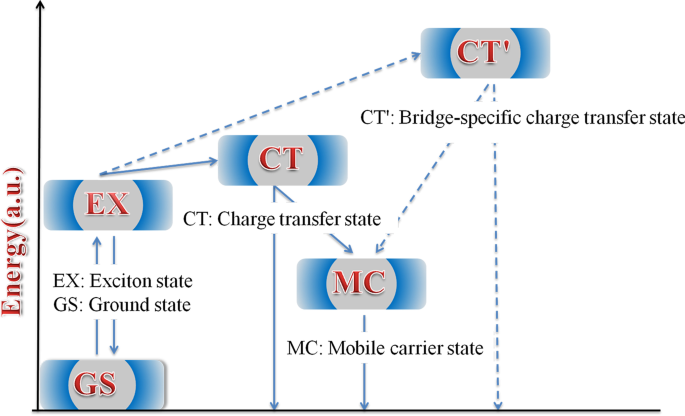
Ilustração esquemática dos estados e caminhos na dinâmica de divisão de excitons dos polímeros HSD em solução
Conclusões
No balanço, usamos uma combinação de estado estacionário e espectroscopia de absorção transiente para estudar o impacto das pontes π em copolímeros de HSD. Verificou-se que a adição da unidade de tiofeno como ponte π levará ao desvio para o vermelho do espectro de absorção em estado estacionário. Enquanto isso, os dados de absorção transiente indicam que HSD-B e HSD-C com unidade de tiofeno como ponte π tinham um estado CT ′ adicional com o pico de impressão digital TA em 1200 nm, que adiciona um novo canal de dissociação de exciton para polímeros HSD. A presença do estado CT ′ torna o polímero vantajoso para a conversão fotoelétrica. Dentre os três polímeros HSD estudados neste trabalho, apenas HSD-B e HSD-C contendo ponte π possuem estados CT ′. Portanto, acreditamos que a existência de pontes π promove a geração de estados CT ′. No entanto, a ponte π não pode ser muito longa. Por exemplo, o polímero HSD-C com dois tiofenos como a ponte π resultando em recombinação não gongeminada devido à existência da ponte π muito longa e afeta a proporção dos estados CT ′. Além disso, também elucidamos as vias de relaxamento do exciton ao analisar o ajuste dinâmico de todas as características espectrais transitórias. Essas descobertas fornecem informações fotofísicas importantes para melhorar a eficiência de conversão de energia dos polímeros conjugados e o desenvolvimento de células solares orgânicas.
Disponibilidade de dados e materiais
Os conjuntos de dados usados e analisados no presente estudo podem ser obtidos dos autores correspondentes mediante solicitação razoável.
Abreviações
- D-π-A:
-
Doador-π-aceitante
- TA:
-
Absorção transitória
- CT ′:
-
Estado de transferência de carga específico da ponte
- OPV:
-
Fotovoltaico orgânico
- PCE:
-
Eficiências de conversão de energia
- NIR:
-
Próximo ao infravermelho
- BHJ:
-
Heterojunção em massa
- TOPAS:
-
Amplificador óptico paramétrico
- WLC:
-
Continum de luz branca
- PL:
-
Fotoluminescência
- VIS:
-
Visível
- GSB:
-
Branqueamento do estado fundamental
- ESA:
-
Absorção de estado de excitação
- EADS:
-
Espectros de diferença associados à evolução
- EX:
-
Estado de excitação
- CT:
-
Estado de transferência de carga
- CS:
-
Estado separado de carga
- MC:
-
Estado da operadora de celular
- SE:
-
Emissão estimulada
Melhoria da ciclabilidade do ânodo de metal de lítio por meio da construção de um canal de íons interlamelar atômico para bateria de enxofre de lítio
Eletrodo de óxido de metal aprimorado para células solares CIGS:a aplicação de uma camada de umedecimento AgOX
Nanomateriais
- O que é corrente alternada (CA)?
- Códigos de identificação da resina
- Polímeros estirênicos renováveis lançados
- Detectando Exciton Espacialmente Localizada em Superredes de Pontos Quânticos Auto-Organizados InAs / InGaAs:Uma Forma de Melhorar a Eficiência Fotovoltaica
- Fabricação, caracterização e citotoxicidade de nanopartículas de carbonato de cálcio derivadas de casca de ouro conjugada em forma esférica para aplicações biomédicas
- Identificação de macromoléculas características de genótipos de Escherichia coli por microscópio de força atômica Mapeamento mecânico em nanoescala
- Preparação de Nanogéis Poliméricos Termorresponsivos de Oligo (Etilenoglicol) Diacrilato-Ácido Metacrílico e Caracterização de Suas Propriedades
- Polímeros piezoelétricos
- Abordagem química para eletrônicos macios mais robustos
- Usinagem CNC de polímeros