


Dependência da Composição das Propriedades Estruturais e Eletrônicas do InGaNBi Quaternário
Resumo
Para realizar engenharia de estrutura de banda viável e, portanto, maior eficiência de luminescência, InGaNBi é uma liga atraente que pode ser explorada em dispositivos fotônicos de luz visível e infravermelho médio. No presente estudo, as propriedades estruturais e eletrônicas como bandgap, energia de divisão spin-órbita e deformação do substrato das composições InGaNBi versus In e Bi são estudadas usando cálculos de primeiros princípios. Os parâmetros de rede aumentam quase linearmente com o aumento das composições In e Bi. Por dopagem com bismuto, o bandgap quaternário do InGaNBi poderia cobrir uma ampla faixa de energia de 3,273 a 0,651 eV para Bi até 9,375% e In até 50%, correspondendo à faixa de comprimento de onda de 0,38-1,9 µm. A energia de divisão spin-órbita calculada é de cerca de 0,220 eV para 3,125%, 0,360 eV para 6,25% e 0,600 eV para 9,375% Bi, respectivamente. Também mostramos a cepa de InGaNBi em GaN; indica que, por meio do ajuste das composições In e Bi, o InGaNBi pode ser projetado em GaN com uma cepa aceitável.
Introdução
Nos últimos anos, wurtzite (WZ) Em x Ga 1− x Ligas N e poços quânticos InGaN / GaN (QWs) têm despertado grande atenção devido ao seu grande potencial para o desenvolvimento de células solares, diodos emissores de luz de alta eficiência (LEDs) e diodos laser (LDs) [1-10]. O comumente usado [0001] -orientado In x Ga 1− x N / GaN QWs sofrem um intenso campo elétrico embutido induzido por estresse compressivo biaxial do In x Ga 1− x Camada N [11], que dá origem à diminuição da energia de emissão QW e da força do oscilador dos pares elétron-buraco. Além disso, existe uma alta densidade de defeitos geométricos em In x Ga 1− x N ligas, incluindo falhas de empilhamento e deslocamentos de rosca (TDs) [12]; esses DTs têm uma grande correlação com centros de recombinação não radiativa. Defeitos, vazamento de elétrons e recombinação Auger são as três fontes para a queda da eficiência de In x Ga 1− x N LEDs, dos quais a recombinação Auger é a principal causa [13].
Da mesma forma, para diodos infravermelhos baseados em GaAs, já foi proposto que a liga de bismuto é um método eficaz para diminuir o bandgap ( E g ), bem como melhorar a divisão spin-orbit (SO) para alcançar a supressão do processo de recombinação Auger [14]. O maior elemento do grupo V do bismuto revela efeitos atraentes nas propriedades físicas das ligas de bismeto. As mudanças na estrutura da banda de ligas de bismeto foram investigadas para diferentes materiais de ligas ternárias tanto experimentalmente quanto teoricamente, como AlNBi [15], GaNBi [16, 17], GaSbBi [18, 19], InPBi [20, 21], e InSbBi [19, 22-24]. O bandgap é modificado principalmente pela grande cepa induzida por átomos de Bi em alta concentração em InPBi. A incorporação de Bi perturba as bandas de valência (VBs) devido à interação dos estados de impureza Bi com bandas de buraco pesado / leve e bandas de separação spin-órbita [21]. Mais recentemente, ligas de bismeto quaternário (por exemplo, GaAsNBi [25-27], InGaAsBi [28, 29], GaAsPBi [30]) também receberam grande atenção. As distorções locais em torno dos átomos P e Bi contribuem significativamente para a modificação do bandgap de GaAsPBi. Um requisito de composição para Ga As 1− x - y P y Bi x para atingir a razão de recombinação Auger mais baixa do que GaAs foi obtida [30]. A combinação de bismuto e outro átomo III ou V aumenta o escopo da engenharia de estrutura de banda, incluindo controle de bandgap, divisão spin-órbita, condução (CB) e desvios de banda de valência e tensão [25]. Portanto, é de interesse significativo descrever o efeito da substituição de Bi no [0001] In x Ga 1− x N / GaN, ajustando as propriedades estruturais e eletrônicas e, portanto, a eficiência da luminescência. No presente estudo, usando cálculos de primeiros princípios [31], as propriedades eletrônicas estruturais, como bandgap, energia de divisão spin-órbita ( Δ SO ), e a cepa de substrato de composições InGaNBi versus In e Bi são estudadas. Considerando a grande incompatibilidade de rede e a baixa qualidade para conteúdo de In superior a 55-60% na amostra InGaN [32], bem como a baixa solubilidade do bismuto em ligas de bismeto diluído, as concentrações de In e Bi são controladas em até 50% e 9,375%, respectivamente. O artigo está organizado da seguinte forma. Na seção “Métodos”, apresentamos os métodos computacionais detalhados. As propriedades estruturais, eletrônicas e deformação do substrato são fornecidas na seção “Resultados e discussão”. Finalmente, um breve resumo é resumido.
Métodos
Nossos cálculos teóricos são baseados na teoria do funcional da densidade (DFT) [31] conforme implementada no código VASP [33, 34]. Nos cálculos das propriedades estruturais, as interações elétron-íon e troca-correlação são tratadas com o método de onda aumentada do projetor (PAW) [35, 36] e a aproximação de gradiente generalizado (GGA) do Perdew-Burke-Ernzerhof (PBE) [37], respectivamente. As configurações de valência-elétron para átomos In, Ga, N e Bi são empregadas como 4 d 10 5 s 2 5 p 1 , 3 d 10 4 s 2 4 p 1 , 2 s 2 2 p 3 , e 5 d 10 6 s 2 6 p 3 , respectivamente. A fim de superar a subestimação do potencial PBE no bandgap das propriedades eletrônicas, empregamos o potencial de troca de Becke-Johnson modificado em combinação com a correlação de aproximação de densidade local (MBJLDA) [38]. O bismuto tem um grande efeito de acoplamento spin-órbita (SOC) e, portanto, o SOC está incluído nos cálculos eletrônicos. Em todos os cálculos, as estruturas são relaxadas até que as forças em cada átomo se tornem menores que 0,02 eV / Å e a mudança máxima de energia seja da ordem de 10 −4 eV. Um corte de onda plana de 450 eV é definido para garantir a precisão dos cálculos. Um Monkhorst-Pack de 4 × 4 × 4 k A malha de pontos é adotada na primeira zona de Brillouin.
Resultados e discussão
Propriedades Estruturais
As supercélulas consistem em 4 × 2 × 2 de célula primitiva WZ-GaN, incluindo 64 átomos. Investigamos 36 composições de I n y Ga 1− y N 1− x Bi x com 0≤ x ≤0,09375,0≤ y ≤0,5 com base em experimentos recentes em que a amostra InGaN exibe grande incompatibilidade de rede e baixa qualidade para conteúdo de In superior a 55-60% [32], bem como a baixa solubilidade de bismuto em ligas de bismeto diluído. Uma configuração representativa é considerada onde os átomos In e Bi estão uniformemente espalhados. Resumimos os parâmetros de rede calculados de ternário In y Ga 1− y N e quaternário Em y Ga 1− y N 1− x Bi x ligas juntamente com outros dados teóricos e experimentais na Fig. 1. Para GaN primitivo, os parâmetros de rede a =3.211, c =5,235 Å, que estão em boa concordância com outros cálculos teóricos a =3,155,3,22 Å, c =5,144,5,24 Å [39-41] e dados experimentais 3,19 Å para a , 5,19 Å para c [42]. Os parâmetros de rede ( a , c ) de Em y Ga 1− y N aumenta quando a composição é aumentada e mostra uma variação quase linear, como mostrado na Fig. 1a. Os cálculos atuais prevêem a =3,304 Å, c =5,365 Å para In 0,25 GaN e a =3,397 Å, c =5,509 Å para In 0,5 GaN, todos os quais concordam bem com os resultados anteriores de a =3,33 Å, c =5,39 Å para In 0,25 GaN e a =3,43,3,485 Å, c =5,55,5,488 Å para I n 0,5 GaN [39, 40, 43, 44]. No caso de ligas quaternárias In y Ga 1− y N 1− x Bi x , no que nos diz respeito, não existem valores experimentais e teóricos para propriedades estruturais. Na Fig. 1b, os parâmetros de rede obtidos também aumentam quase linearmente com o aumento das composições In e Bi. Por causa dos raios iônicos maiores de In e Bi do que os átomos de Ga e N, a substituição de In sobre Ga e Bi sobre N leva a parâmetros de rede aprimorados de InGaNBi.
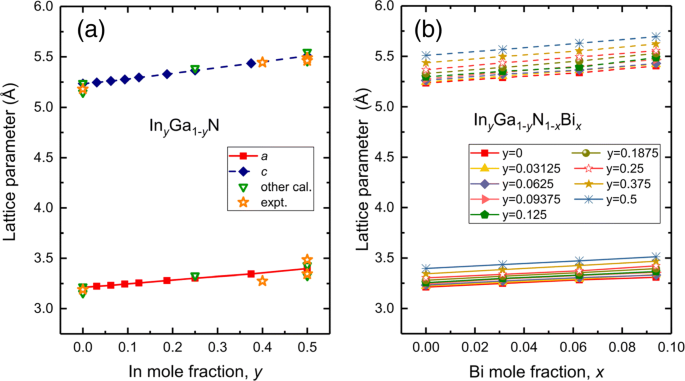
Os parâmetros de rede para a ligas ternárias In y Ga 1− y N , com 0≤ y ≤0,5 e b ligas quaternárias Em y Ga 1− y N 1− x Bi x , com 0≤ x ≤0,09375, 0≤ y ≤0,5. Para comparação, adicionamos alguns outros cálculos e dados experimentais da Ref. [39–44] na Fig. 1a. A linha sólida representa a e a linha tracejada é c
A incorporação de In e Bi quebrará a periodicidade do cristal e introduzirá deformação geométrica em uma estrutura fortemente ligada. Nós escolhemos Em 0,25 GaN Bi 0,0625 como um exemplo para quatro estatísticas de ligações químicas, como mostrado na Fig. 2; os comprimentos médios das ligações Ga-N, In-N, Ga-Bi e In-Bi são 2,009, 2,195, 2,592 e 2,704 Å, respectivamente. Observe que o comprimento da ligação Ga-N no GaN original é 1,970 Å. O comprimento da ligação In-N é maior do que o de Ga-N, o que indica que o átomo In empurra acentuadamente o átomo N para longe. Da mesma forma, o comprimento da ligação maior de Ga-Bi do que Ga-N significa que o átomo de Bi empurra o átomo de Ga para longe, encontrando boa consistência com a ordem dos raios covalentes de Ga (1,22 Å), In (1,42 Å), N (0,71 Å), e Bi (1,48 Å) [45]. Outras configurações exibem comportamento semelhante. A deformação reticulada e a disparidade na eletronegatividade entre o hospedeiro e o dopante têm um efeito considerável nas propriedades eletrônicas e ópticas.
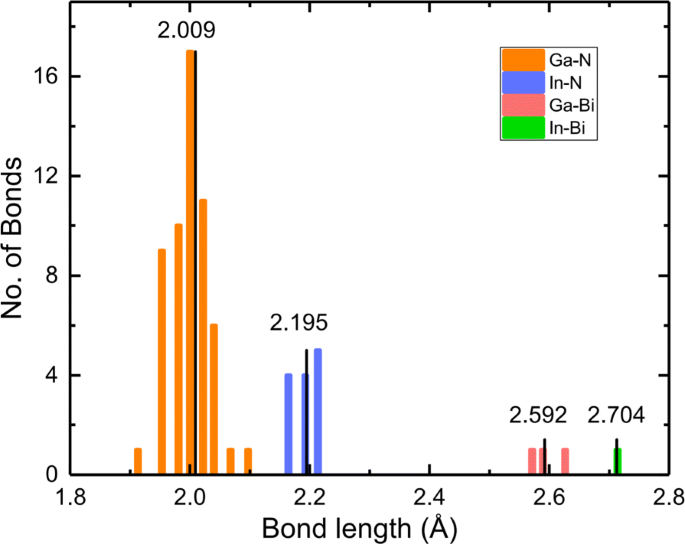
Histograma do comprimento da ligação em In 0,25 GaNBi 0,0625 . Os valores no painel indicam os comprimentos médios dos quatro tipos de títulos
Propriedades eletrônicas
Foi demonstrado que os potenciais funcionais ou de correção e o efeito SOC influenciam muito a precisão prevista da energia do bandgap da liga III-V, da banda de valência e da energia de divisão spin-órbita. Assim, validamos nossos resultados usando o potencial MBJLDA e comparamos com outros cálculos teóricos e experimentos. A Figura 3 é um gráfico da energia do bandgap em relação à composição em In y Ga 1− y N, bem como um ajuste aos dados. Alguns valores de bandgap obtidos por experimentos, funcionais teóricos HSE06, mBJ e LMTO-CPA-MBJ também são plotados. O bandgap previsto de GaN é 3,273 eV, que está em boa consistência com os cálculos e experimentos atuais, 3,33 eV por mBJ [40], 3,261, 3,23 eV por HSE06 [39, 46], e 3,40-3,50 eV por experimentos [47- 49]. Conforme observado em I n y G a 1− y N, nossos resultados DFT confirmam que E g valores de eu n y G a 1− y N diminui continuamente conforme y é aumentado de 0 a 50%. E g diminui suavemente de 3,273 para 1,546 eV. Isso se compara bem com os resultados teóricos (potenciais HSE06, mBJ) [39, 40, 46] e experimentais [50, 51].
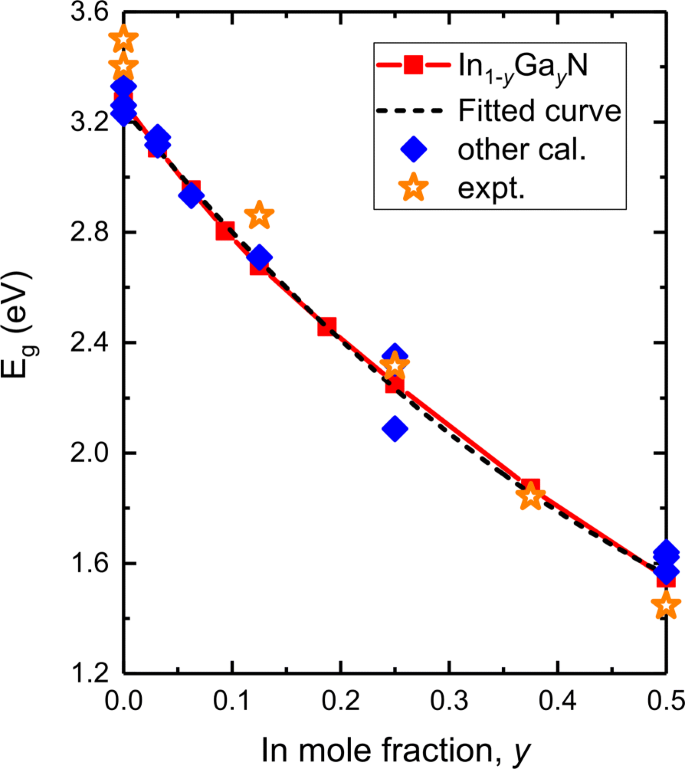
Energia prevista de bandgap ( E g , linha sólida vermelha) como uma função de Na composição em I n y G a 1− y N bem como um ajuste aos dados (linha tracejada preta). Outros resultados teóricos [39, 40, 46] e experimentais [47-51] também são plotados
O gráfico de contorno para o bandgap do quaternário I n y G a 1− y N 1− x B eu x ligas é mostrado na Fig. 4. Os bandgaps das ligas quaternárias exibem uma tendência não linear em função da composição, que diminui com o aumento dos teores de In e Bi. A partir dos resultados, descobrimos que o bandgap do InGaNBi poderia cobrir uma ampla faixa de energia de 3,273 a 0,651 eV para Bi até 9,375% e In até 50%, correspondendo à faixa de comprimento de onda de 0,38 a 1,9 µm, indicando suas potenciais aplicações optoeletrônicas em luz visível e alcance infravermelho médio.
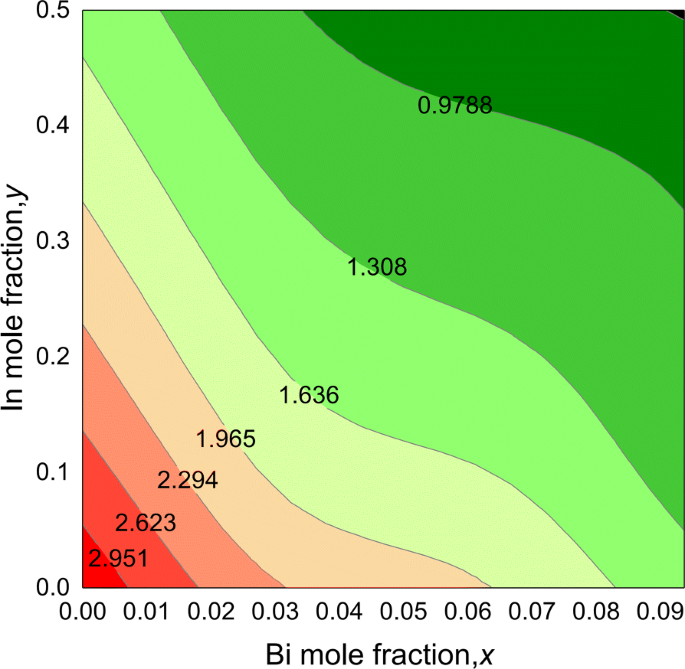
Gráfico de contorno dos valores de bandgap para I n y G a 1− y N 1− x B eu x ligas, em função de Bi ( x ) e In ( y ) composições
Em comparação com InGaN, a incorporação de Bi induz uma redução mais acentuada do bandgap. Mas, além disso, um aumento significativo em Δ SO é obtido devido ao forte efeito SOC do bismuto onde a interação avançada entre o spin do elétron e o momento angular orbital diminui a energia da banda SO. Além disso, a borda da banda de valência melhorada originada do efeito anti-cruzamento da banda de valência das ligas de bismeto também melhora amplamente Δ SO [28]. Nosso calculado Δ SO os valores são cerca de 0,220 eV para 3,125%, 0,360 eV para 6,25% e 0,600 eV para 9,375% Bi, respectivamente, o que tem uma variação insignificante com a fração de índio. Investigações anteriores demonstraram que diferentes arranjos Bi são de grande influência nas estruturas de bandas de ligas de bismeto, incluindo a energia de divisão spin-órbita [21, 52]. Os resultados atuais mostram que o eu n 0,5 G a N B eu 0,09375 o valor do bandgap (0,651 eV) é muito próximo ao de Δ SO (0,577 eV). Uma vez que a amostra InGaN exibe grande incompatibilidade de rede e baixa qualidade para conteúdo de In superior a 55-60% [32], bem como a baixa solubilidade de bismuto em ligas de bismeto diluído, definimos os conteúdos de In até 50% e Bi até 9,375%. Acreditamos que um conteúdo mais alto de índio ou bismuto alcançará Δ SO > E g em amostra InGaNBi quaternária para aumentar a eficiência de LEDs e LDs baseados em InGaNBi.
As estruturas de banda projetadas e a densidade total de estados (TDOS) de GaN primitivo, I n 0,25 GaN e eu n 0,25 G a N B eu 0,03125 as ligas são apresentadas na Fig. 5. As contribuições de In e Bi são destacadas pela cor:azul (vermelho) corresponde ao estado originado de In (Bi). A substituição em I n 0,25 O GaN tem grande influência na banda de condução e na banda de valência, onde o mínimo da banda de condução (CBM) é empurrado para energias mais baixas em relação ao nível de Fermi e refletem o gap de energia mais estreito. Ao contrário do bismuto que introduz a banda de defeito na lacuna proibida perto do nível de Fermi, os átomos de In mostram uma hibridização com o nível profundo do VB. Para liga quaternária I n 0,25 G a N B eu 0,03125 , pode ser visto claramente que a redução dos resultados de bandgap tanto do máximo da banda de valência ascendente (VBM) quanto do CBM descendente, e o CBM muda mais significativamente em comparação com I n 0,25 GaN, que é atribuído à maior deformação compressiva em InGaNBi pela adição de bismuto. O nível de defeito destacado pela cor vermelha tem uma forte interação com a borda VB, que é derivada da hibridização principalmente entre os átomos Bi e próximos de Ga. O TDOS na Fig. 5e também reflete o nível de defeito local em -1,0 a -0,5 eV.
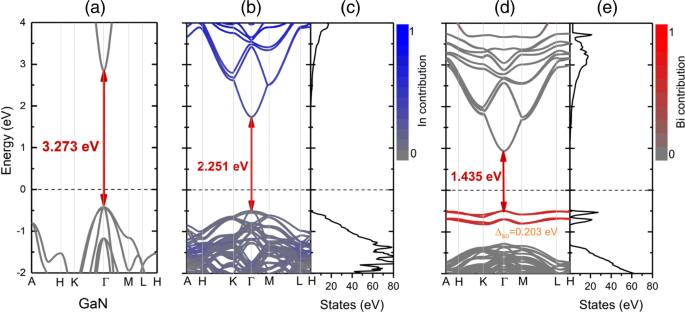
As estruturas de banda projetadas e suas correspondentes densidade total de estados (TDOS) de a GaN, b , c eu n 0,25 G a N , e d , e eu n 0,25 G a N B eu 0,03125 . A linha tracejada preta representa o nível de Fermi, que é definido como zero. As contribuições relativas de In e Bi são destacadas pela cor:azul (vermelho) corresponde ao estado originário de In (Bi)
Estirpe de InGaNBi em GaN
O [0001] -orientado I n y G a 1− y Os poços quânticos tensos N / GaN são amplamente adotados nos dispositivos atuais de LED e LD, nos quais I n y G a 1− y Camadas N sofrem uma tensão compressiva biaxial. Flutuações composicionais locais e diferentes raios covalentes de átomos de In e Ga dão origem às cepas em I n y G a 1− y Camadas N [53]. A Figura 6 mostra a cepa de InGaNBi em um substrato de GaN. Como o átomo de índio é maior do que o átomo de gálio, o átomo de bismuto é maior do que o átomo de nitrogênio; assim, incorporar átomos In e Bi em InGaNBi induz a tensão compressiva InGaNBi em GaN. É mostrado que no teor de In de 50% e teor de Bi de 9,375%, InGaNBi está sob alta deformação compressiva de 8,5%. Para a fração In dentro de 6,25% e a fração Bi dentro de 2,8%, a cepa de InGaNBi em GaN está dentro de 1%. Ou seja, por meio do ajuste das composições In e Bi, o InGaNBi pode ser projetado em GaN com uma deformação aceitável.
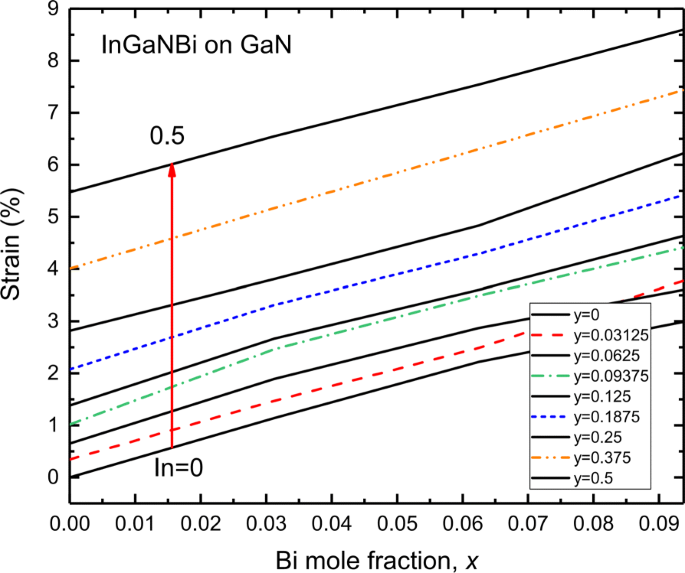
Deformação de ligas de InGaNBi em substrato de GaN em vários In (0–0,5) como uma função da fração Bi. Valores positivos de deformação indicam que InGaNBi está sob deformação compressiva
Conclusões
As propriedades estruturais, eletrônicas e deformação de InGaNBi em composições de GaN versus In e Bi são investigadas com base na teoria do funcional de densidade. Os parâmetros de rede do InGaNBi aumentam quase linearmente com o aumento das composições In e Bi. Como os átomos In e Bi têm o raio atômico maior do que os átomos Ga e N, os comprimentos das ligações In-N e Ga-Bi são maiores do que os de Ga-N. Para propriedades eletrônicas, mostramos o gráfico de contorno para o bandgap do quaternário I n y G a 1− y N 1− x B eu x ligas. O bandgap das ligas quaternárias poderia cobrir uma ampla faixa de energia de 3,273 a 0,651 eV para Bi até 9,375% e In até 50%, correspondendo à faixa de comprimento de onda de 0,38 a 1,9 µm. O Δ calculado SO os valores são cerca de 0,220 eV para 3,125%, 0,360 eV para 6,25% e 0,600 eV para 9,375% Bi, respectivamente, o que tem uma variação insignificante com a fração de índio. Acreditamos que uma composição mais elevada de índio ou bismuto alcançará Δ SO > E g em amostra InGaNBi quaternária para aumentar a eficiência de LEDs e LDs baseados em InGaNBi. A análise da estrutura de bandas mostra que o índio tem grande influência tanto no CB quanto no VB, e o bismuto tem uma forte interação com a borda do VB. Finalmente, investigamos a cepa de InGaNBi em GaN. Através do ajuste das composições In e Bi, InGaNBi pode ser projetado em GaN com uma deformação aceitável.
Influência de Te-Doping em Nanowires VS InAs sem Catalisador
Um Memristor multinível baseado em filme fino HfO2 Al-Dopado
Nanomateriais
- PPA Reforçado com Fibra de Carbono para Componentes Automotivos Estruturais e Eletrônicos
- Estrutura e propriedades eletrônicas da nanoargila caulinita dopada com metal de transição
- Modulação das propriedades de anisotropia eletrônica e óptica de ML-GaS por campo elétrico vertical
- O efeito do plasma sem equilíbrio de contato nas propriedades estruturais e magnéticas de Mn Х Fe3 - X О4 Spinels
- Propriedades ópticas e eletrônicas de fotodiodos N + / P de silício hiperdopado com enxofre induzido por laser de femtosegundo
- Propriedades ópticas estruturais e de infravermelho próximo visível de TiO2 dopado com Cr para pigmentos frios coloridos
- Sondando as propriedades estruturais, eletrônicas e magnéticas dos aglomerados Ag n V (n =1–12)
- Dependência da toxicidade das nanopartículas em suas propriedades físicas e químicas
- Propriedades de PCB automotivo e considerações de design
- Propriedades e Composição do Ferro Gusa