


Relações lógicas e dependentes de elétrons de valência fortes de impurezas elementares em semicondutor binário 2D:um caso de monocamada GeP3 de estudos de Ab Initio
Resumo
Usando cálculos de primeiro princípio dentro da teoria do funcional da densidade, investigamos a propriedade eletrônica e a estabilidade de GeP 2D com dopagem substitutiva 3 monocamada com dopantes do grupo III a VI. As propriedades de condução são dramaticamente modificadas pelos locais de dopagem e pelo número de elétrons de valência dos dopantes. Especificamente, a substituição no local Ge exibe oscilações de metal-semicondutor em função do número de elétrons de valência dos dopantes, enquanto tais oscilações são totalmente revertidas quando a substituição no local P. Além disso, também estudamos o caso de co-doping em GeP 3 , mostrando que o co-doping pode produzir um fenômeno lógico “AND”, ou seja, as propriedades condutoras do GeP co-dopado 3 pode ser deduzida por meio de uma relação lógica simples de acordo com os resultados do doping único. Finalmente, investigamos a energia de formação de dopantes e descobrimos que os sistemas co-dopados elétron-buraco e buraco-buraco são muito mais energeticamente favoráveis devido à atração de Coulomb. Nossos resultados não apenas apresentam uma compreensão abrangente do fenômeno de dopagem 2D, mas também propõem uma rota intrigante para ajustar as propriedades eletrônicas de semicondutores binários 2D.
Introdução
Desde a descoberta do grafeno [1, 2], a família de cristais bidimensionais (2D), como dichalcogenetos de metais de transição (TMDs) [3], siliceno [4], germaneno [5], fosforeno [6], telureno [ 7], e assim por diante, têm atraído grande atenção devido às suas propriedades elétricas, ópticas e magnéticas únicas [8,9,10]. Por exemplo, o grafeno se comporta como férmions de Dirac sem massa, o que dá origem à mobilidade final dos portadores de alta carga [11, 12]. Assim, é promissor para apoiar o efeito Hall de spin quântico 2D, termoeletricidade aumentada, supercondutividade [13], e até mesmo o efeito Hall anômalo quântico [14,15,16]. Combinado com o número crescente de bancos de dados de estrutura de cristal disponíveis, ferramentas computacionais modernas têm sido usadas para explorar materiais 2D recentemente desconhecidos. Até agora, mais de 1000 materiais 2D são previstos e alguns deles são fabricados em experimentos [17,18,19], tornando-se um campo interessante na física, química e ciência dos materiais. Tais estudos fundamentais e explorações de materiais 2D também aumentam suas grandes aplicações potenciais para o campo de detecção [20,21,22,23,24,25].
Recentemente, Jing et al. relatou novo material 2D-GeP 3 monocamada, que tem maior estabilidade química do que a monocamada BP e possui excelentes propriedades eletrônicas e ópticas. Além disso, o 2D GeP 3 monocamada parece ter uma propriedade semicondutora devido ao forte confinamento quântico entre camadas. Eles descobriram que o GeP 3 monocamada exibe um gap moderado e ajustável de cerca de 0,55 eV [26]. Com base na alta capacidade e boa estabilidade cíclica, GeP 3 filme fino é proposto para baterias de íon-lítio como um ânodo promissor [27]. Li et al. também investigou o GeP 3 nanoribbon e descobriu que as lacunas de banda podem exibir oscilações pares-ímpares com o aumento da largura [28].
O doping é uma estratégia prática para ajustar fundamentalmente as propriedades eletrônicas e magnéticas dos materiais hospedeiros em camadas 2D [29]. Além disso, quebra a limitação de um único material nas aplicações de diversos campos e dispositivos. Como sabemos, o semicondutor de monocamada 2D pode resultar em interações elétron-elétron notavelmente aprimoradas que foram demonstradas para gerar renormalização de grande intervalo de banda e exciton de ambos os cálculos teóricos de muitos corpos e experimentos [30, 31]. Comparado com o doping em semicondutores a granel, o doping em 2D também deve exibir alguns comportamentos anormais devido ao forte efeito de confinamento de elétrons, ou seja, grafeno dopado com boro ou nitrogênio é possível abrir um pequeno gap no ponto de Dirac, e o A lacuna de banda do grafeno também pode ser aberta efetivamente em torno de pontos K (ou K ') pela introdução de pequenos domínios BN [32]. As lacunas de banda de fosforeno preto mostram um comportamento oscilante por dopagem de diferentes elementos com números pares ou ímpares de elétrons de valência [33, 34]. Neste trabalho, tentamos estender a investigação de elementos de dopagem do grupo IV – V em GeP binário 2D 3 semicondutor monocamada.
Aqui, realizamos os estudos sistemáticos do GeP dopado substitucionalmente 3 monocamada com os dopantes do grupo III a VI. As propriedades eletrônicas dos sistemas dopados serão dramaticamente afetadas pelo número de elétrons de valência dos dopantes e pelos locais de dopagem. Os grãos centrais são (1) para dopante único, os resultados são sensivelmente dependentes dos locais de substituição e a substituição em dois tipos de locais de dopagem produzirá resultados totalmente inversos. (2) As propriedades condutoras de co-dopagem podem ser deduzidas por um operador lógico através das do dopante único. Além disso, a energia de formação calculada de diferentes tipos de dopagem sugere que alguns deles são altamente favoráveis energeticamente contra a flutuação térmica.
Métodos Computacionais
Todos os nossos cálculos da teoria funcional da densidade dentro da aproximação do gradiente geral são realizados usando o Vienna ab initio Simulation Package [35]. Os termos de troca e correlação foram descritos com o funcional Perdew-Burke-Ernzerhof (PBE), e o potencial de onda aumentada do projetor foi empregado para descrever a interação elétron-íon [36,37,38]. O GeP dopado 3 monocamada foi modelada em uma supercélula 2 × 2 periódica contendo 32 átomos, e uma supercélula maior de 3 × 3 também foi usada para verificar nossos resultados. Um espaço de vácuo de cerca de 20 Å ao longo do z direção foi adotada a fim de eliminar a interação entre as camadas vizinhas. Para a dopagem simples, um átomo de Ge ou P foi substituído por um dopante do grupo III (IV, V e VI). As estruturas geométricas são determinadas pela comparação com os resultados relatados, incluindo constante de rede e propriedade eletrônica do host GeP 3 monocamada. Nos sistemas de dopagem, todos os átomos nas supercélulas podem relaxar até que a força Hellmann-Feynman seja inferior a 0,02 eVÅ −1 , mas as constantes de rede das células da superfície são fixadas durante o relaxamento do átomo. Um corte de energia cinética de cerca de 600 eV e 6 × 6 × 1 k - malhas foram usadas, respectivamente [39].
Para verificar a disponibilidade dos dopantes no GeP 3 monocamada, a energia de formação ( E f ) de dopantes X ( X =grupo III – VI) é calculado de acordo com as duas fórmulas a seguir. Para dopante único, temos o seguinte:
$$ {\ mathrm {E}} _ {\ mathrm {f}} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3:\ mathrm {X} \ right) =\ mathrm {E} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3:\ mathrm {X} \ right) - \ mathrm {E} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3 \ right) - { E} _ {\ mathrm {X}} + {E} _ {\ mathrm {i}} $$ (1)
e para o sistema de co-dopagem, uma fórmula semelhante é usada:
$$ {\ mathrm {E}} _ {\ mathrm {f}} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3:\ mathrm {XY} \ right) =\ mathrm {E} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3:\ mathrm {XY} \ right) - \ mathrm {E} \ left (\ mathrm {Ge} {\ mathrm {P}} _ 3 \ right) - { E} _ {\ mathrm {X}} - {E} _ {\ mathrm {Y}} + {E} _ {\ mathrm {i}} + {E} _ {\ mathrm {j}} $$ (2 )
onde E f (GeP 3 : X ) e E (GeP 3 ) são as energias totais do GeP dopado com X e intrínseco 3 monocamada com a mesma supercélula. E (GeP 3 : XY ) São as energias totais do sistema co-dopado XY, E X e E Y são as energias atômicas dos dopantes X ou Y referentes às suas estruturas em massa correspondentes, e E i , E j são as energias dos átomos substituídos onde i e j indicam o átomo Ge ou P, respectivamente [40, 41].
Resultados e discussões
Oscilações pares-ímpares para sistemas de dopagem de elemento único
A Figura 1a mostra a vista superior e lateral da estrutura do GeP 3 2 × 2 supercélula, e a Fig. 1b é a zona de Brillouin 2D correspondente de GeP 3 monocamada. As constantes de rede otimizadas de GeP 3 monocamada são \ (\ mathrm {a} =\ mathrm {b} =6,96 \ {\ AA} \), e o intervalo de banda calculado é de cerca de 0,26 eV, o que está de acordo com outros cálculos teóricos.
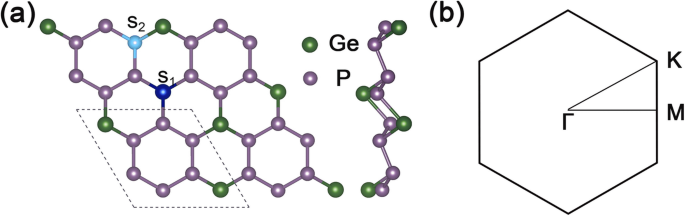
Estrutura geométrica e zona Brillouin de GeP 3 . a Vistas superior e lateral da geometria otimizada de GeP 3 com uma supercélula 2 × 2. A linha pontilhada apresenta a célula unitária de GeP 3 monocamada, S 1 representa o local de substituição da posição do local Ge, e S 2 representa o local de substituição do átomo de posição P. b a zona 2D Brillouin de GeP 3 monocamada
Em primeiro lugar, traçamos as estruturas de banda de GeP dopado com um único elemento 3 monocamada com um átomo de Ge substituto (aqui, escolhemos B, C, N, O, Al, Si, P, S, Ga, As e Se como dopantes). Os resultados são mostrados na Fig. 2a – l, respectivamente. Podemos ver claramente que o nível de Fermi sobe e cruza as bandas de condução para o grupo V (N, P, As) devido a mais um dopante de elétron, enquanto que para o grupo III (B, Al, Ga) dopante devido a um elétron a menos, o um desloca para baixo e cruza as bandas de valência. Por exemplo, nas Fig. 2f e j, o máximo de sua banda de valência corresponde apenas às bandas parcialmente ocupadas mostradas nas Fig. 2e e i. No entanto, para dopantes do grupo IV (C, Si e Ge) e VI (O, S e Se), por causa do mesmo ou mais dois elétrons do átomo de Ge, os sistemas apresentam uma característica semicondutora. Essa sintonia da transição semicondutora para metálica deriva da ocupação do número de elétrons de valência, ou seja, a ocupação de elétrons de valência ímpares (pares) leva a propriedades metálicas (semicondutoras).
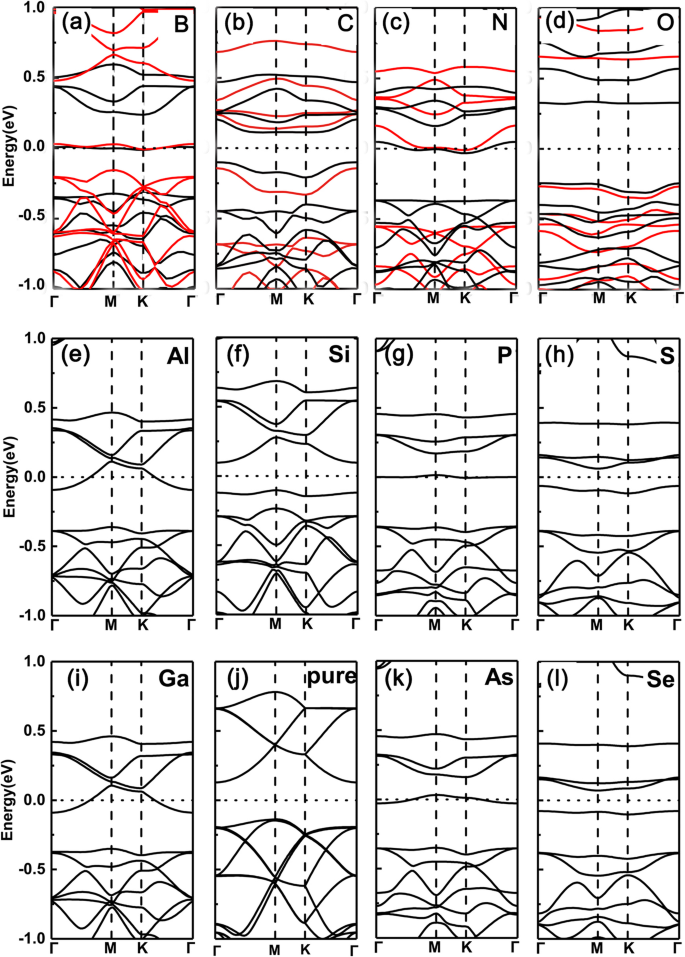
Estruturas de banda dos vários dopantes em GeP 3 monocamada com substituição do átomo de Ge. a B, b C, c N, d O, e Al, f Si, g P, h S, i Ga, j GeP puro 3 , k Como, l Se. Estruturas de banda calculadas para uma supercélula \ (\ mathsf {2} \ times \ mathsf {2} \) com vários dopantes em GeP 3 monocamada do grupo III a VI, com substituição do átomo de Ge, respectivamente, juntamente com o do GeP puro 3 monocamada. Os funcionais PBE e HSE06 são empregados na linha superior
Para confirmar a validade dos resultados acima derivados de funcionais PBE, também empregamos os funcionais de densidade híbrida (HSE06) para verificar os sistemas dopados da linha superior. É claro que os funcionais PBE realmente dão os erros de lacunas de banda devido à subestimação. No entanto, em nossos sistemas estudados, todos eles têm lacunas consideráveis, isso significa que os erros entre propriedades metálicas ou semicondutoras causados pelos funcionais PBE geralmente não acontecerão (Isso ocorre porque em algumas pequenas lacunas de banda de semicondutores, os funcionais PBE geralmente dão origem ao erro entre propriedades condutoras e metálicas). Além disso, em nosso estudo, o que nos preocupa são as características metálicas ou semicondutoras, ao invés dos valores específicos de band gaps. Comparado com as lacunas derivadas de funcionais PBE, as lacunas de funcionais HSE06 aumentam claramente. Mesmo assim, as oscilações do metal semicondutor permanecem intactas. Portanto, os ingredientes centrais desenhados com base nos funcionais PBE são confiáveis.
No entanto, em nítido contraste, os casos de substituição de átomos de P pelos mesmos dopantes são totalmente revertidos, como mostrado na Fig. 3a-l, respectivamente. Ou seja, para os dopantes do grupo V (N, As) e do grupo III (B, Al, Ga), os sistemas dopados permanecem com propriedade semicondutora, enquanto para o grupo IV (C, Si, Ge) e VI (O, S, Se) dopantes, aqueles mudam para característica metálica (aqui, a mesma tendência também é encontrada entre os funcionais PBE e HSE06). Isso ocorre porque os elétrons de valência mantêm o mesmo (dois a menos) que (do que) o GeP intrínseco 3 para dopantes do grupo V (grupo III), mas um elétron a menos (mais) para dopantes do grupo IV (VI).
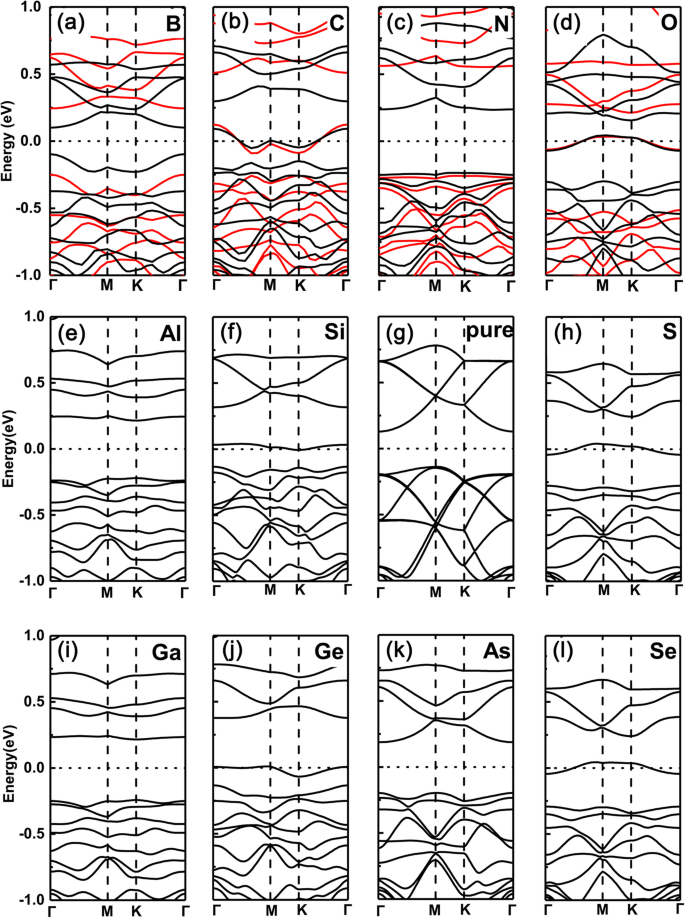
Estruturas de banda dos vários dopantes em GeP 3 monocamada com substituição do átomo de P. a B, b C, c N, d O, e Al, f Si, g P, h S, i Ga, j GeP puro 3 , k Como, l Se. Estruturas de banda calculadas para uma supercélula \ (\ mathsf {2} \ times \ mathsf {2} \) com vários dopantes em GeP 3 monocamada do grupo III a VI, com substituição de átomos de P, respectivamente, juntamente com a de GeP puro 3 monocamada. Os funcionais PBE e HSE06 são empregados na linha superior
Para melhor apresentar as oscilações da transição das propriedades semicondutoras para metálicas, traçamos a tendência de mudança do gap como os diferentes dopantes, conforme mostrado nas Fig. 4a eb, respectivamente. Claramente, podemos ver que a transição das propriedades semicondutoras para as metálicas está se revertendo drasticamente. Especificamente, as oscilações metálicas (semicondutoras) - semicondutoras (metálicas) ocorrem na substituição do local Ge (P) como dopantes que variam do grupo III a VI. Além disso, também encontramos um fenômeno interessante que mostra que a magnitude do bandgap quase se mantém a mesma do GeP intrínseco 3 monocamada quando os dopantes têm os mesmos elétrons de valência que o átomo de Ge. No entanto, quando os dopantes têm dois elétrons a mais do que o átomo de Ge, a magnitude dos intervalos de banda muda relativamente maior. No entanto, para os dopantes em locais P, independentemente do número de elétrons de valência, a magnitude dos intervalos de banda sempre muda relativamente grande. Isso pode ser entendido pelo efeito conjunto do raio do átomo e os elétrons de valência disponíveis, ou seja, dopantes com quase o mesmo (menor ou maior) raio e elétrons de valência que (do que) o átomo de Ge causam um efeito relativamente menor (maior) no sistema eletrônico propriedades, como o gap. Isso significa que é possível ajustar não apenas as oscilações das transições semicondutor-metálicas, mas também a magnitude do gap, escolhendo dopantes adequados e os diferentes locais de dopagem.
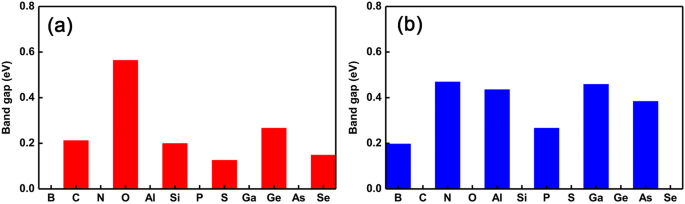
Lacunas de banda de todos os sistemas com dopagem simples. Lacunas de banda de GeP dopado 3 monocamadas com os diferentes dopantes que variam do grupo V a VI. a a substituição de átomos de Ge e b os átomos de substituição P, respectivamente
Para entender a mudança das estruturas eletrônicas de diferentes dopantes no GeP 3 monocamada, traçamos a densidade parcial de estados (PDOS) do grupo intrínseco e de dopagem IV – V em GeP 3 monocamada, conforme mostrado na Fig. 5a – d, respectivamente. Pode ser visto claramente que a banda de valência máxima (VBM) e a banda de condução mínima (CBM) do GeP 3 monocamada origina-se principalmente de orbitais p de átomos de Ge e P. Quando os dopantes com o mesmo número de elétrons de valência que o átomo de Ge, como C e Si, estão disponíveis, haverá estados de impureza apenas localizando acima do VBM do GeP intrínseco 3 monocamada porque o nível de energia orbital p de C e Si é mais alto do que o do átomo P (ver Fig. 5a). Portanto, a propriedade condutora está intacta e a magnitude da mudança do gap é relativamente pequena. No entanto, quando os dopantes têm um elétron a mais do que o átomo de Ge, como N, P e As, também haverá estados de impureza no gap de banda e os estados de impureza originados da hibridização da divisão CBM (dominante) e os estados de dopantes (ver Fig. 5b).
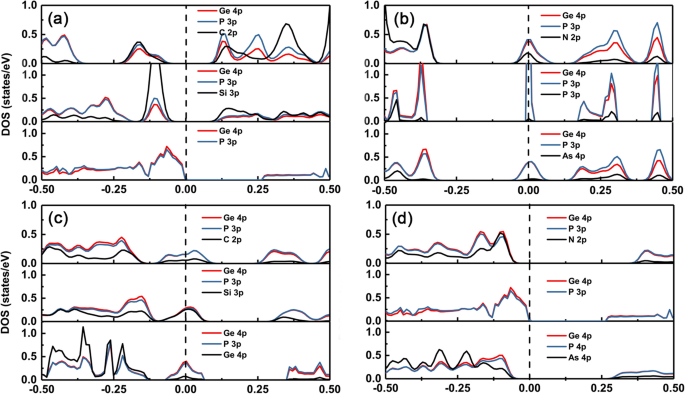
DOS para os sistemas dopados. A densidade parcial de estados (direita) para átomos do grupo IV (C, Si e Ge) e do grupo V (N, P e As) dopados com GeP 3 . A linha tracejada vertical enegrecida é o nível de Fermi. (a) e (b) átomo de Ge substituído, (c) e (d) átomo de P substituído
Pelo contrário, para o doping no sítio P, quando os dopantes têm um elétron de valência a menos do que o átomo P, como o grupo IV, haverá estados de impureza em todo o nível de Fermi, e os estados de impureza são compostos de divisão VBM (dominante ) e os estados dos dopantes. Ao passo que, quando os dopantes têm o mesmo número de elétrons de valência que o átomo P, como o grupo V, os sistemas dopados ainda mantêm uma característica semicondutora (ver Fig. 5c). As lacunas de banda tornam-se relativamente maiores do que aquelas do GeP 3 intrínseco monocamada devido à maior incompatibilidade de constantes de rede. Além disso, também observamos que o gap do dopante N é maior do que aquele do dopante As na substituição de átomos de P. Isso ocorre porque o nível de energia p orbital do átomo As é mais alto do que o do átomo N, portanto, quanto mais alto o nível de energia p orbital, maior é o deslocamento para cima dos estados de impureza para longe do VBM (ver Fig. 5d).
Relações lógicas para sistemas de co-dopagem
Com base nas descobertas acima mencionadas de diferentes dopantes individuais, podemos, portanto, ser capazes de projetar sistemas de co-dopagem para satisfazer as propriedades eletrônicas que desejamos. Aqui, mostramos apenas os resultados de B, C, N e O como exemplos para ilustrar o efeito de co-dopagem, mas a conclusão é robusta contra os diferentes dopantes selecionados. Por exemplo, no site Ge de co-dopagem, ambos os dois dopantes com um elétron de valência a menos podem levar naturalmente à propriedade semicondutora, enquanto para os dois dopantes com um a menos e mais número de elétrons de valência, os sistemas de co-dopagem também podem, portanto, têm uma propriedade semicondutora.
No entanto, para os dois dopantes com um a menos (mais) e o mesmo elétron de valência, os sistemas de co-dopagem ainda mantêm a propriedade metálica como um número a menos (mais) de propriedades resultantes de dopantes de elétrons de valência. Simplifique, esta ideia é exatamente confirmada por nossos cálculos adicionais de teoria funcional de densidade (DFT) de sistemas co-dopados, veja os resultados na Fig. 6a – l para as estruturas de banda de B, C, N e O co-dopado GeP 3 monocamada.
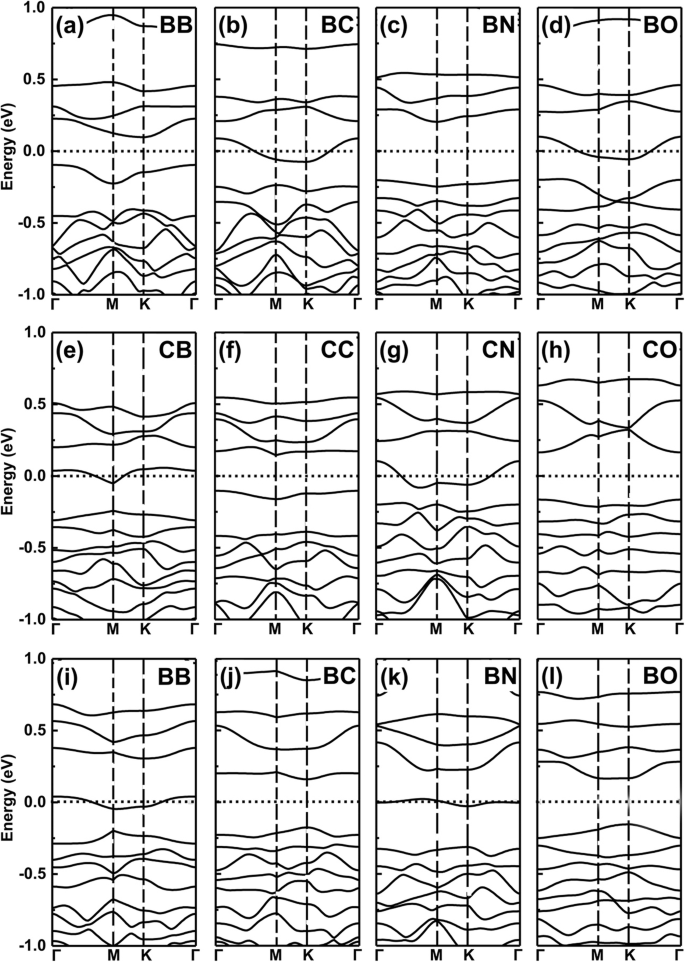
Estruturas de bandas de sistemas co-dopados. As estruturas de banda de B, C, N e O co-dopadas GeP 3 monocamada. a - d Os dois dopantes substituem dois átomos de Ge em GeP 3 monocamada, e - h os dois dopantes substituem dois átomos P, i - l os dois dopantes substituem um átomo de Ge e um átomo de P, respectivamente
Agora, podemos dar um exemplo de operação lógica "AND", definindo a propriedade metálica como "M" e a característica semicondutora como "S". Definimos as relações lógicas:M AND M =S, S AND S =S e M AND S =M, respectivamente. Aqui, esses achados que obtivemos acima obedecem a tais relações lógicas, por exemplo, os dopantes com mais e um menos de elétron de valência resultam em propriedade metálica, mas quando usamos os dois dopantes como co-dopagem, como B e N em locais Ge como mostrado nas Fig. 7a eb, os sistemas co-dopados tornam-se propriedades semicondutoras como esperado, veja a Fig. 6c. Se escolhermos GeP co-dopado com B-C 3 sistema monocamada, apresenta característica metálica que é o caso de M AND S (ver Fig. 4a, b). O mesmo para átomos C-N, N-O e B-O co-dopagem em GeP 3, substituindo dois átomos de Ge, dois átomos de P ou um átomo de Ge e P, como mostrado na Fig. 7c-f, respectivamente.
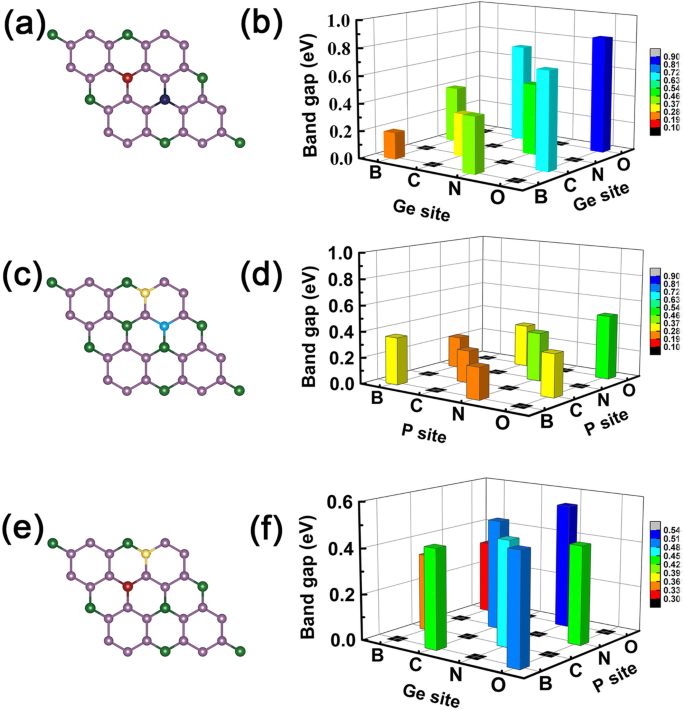
Lacunas de banda de todos os sistemas co-dopados. A magnitude das lacunas de banda de GeP co-dopado 3 monocamada, as esquerdas são o esboço de locais co-dopados e os direitos são a magnitude das lacunas de banda correspondentes aos elementos de dopagem. a , b O caso dos elementos de dopagem ocupam os dois átomos de Ge. c , d O caso dos elementos de dopagem ocupam os dois átomos de P. e , f O caso para elementos de dopagem ocupam os átomos Ge e P, respectivamente
Finalmente, verificamos a estabilidade de ambos os sistemas de dopagem simples e sistemas de dopagem única para garantir se eles podem ser realizados posteriormente no experimento. A energia de formação é calculada usando as Eqs. (1) e (2) para casos de dopagem única e co-dopagem, respectivamente. Os resultados estão presentes na Fig. 8a-e. Da Fig. 8a eb, podemos ver claramente que o E f dos dopantes individuais em locais Ge são todos próximos aos de GeP 3 monocamada (configurada para zero como ponto de referência), exceto átomos C, N e S. Notamos também que, para dopantes de átomos B, O, P, Ge e Se, as energias de formação são muito menores do que outros dopantes, indicando que são muito fáceis de dopar em um experimento. Para dopantes nos sítios P, dopantes de átomos B, O, P e Ge têm energia de formação relativamente menor e também são fáceis de dopar. C, N, Al e Ga não são fáceis para doping.
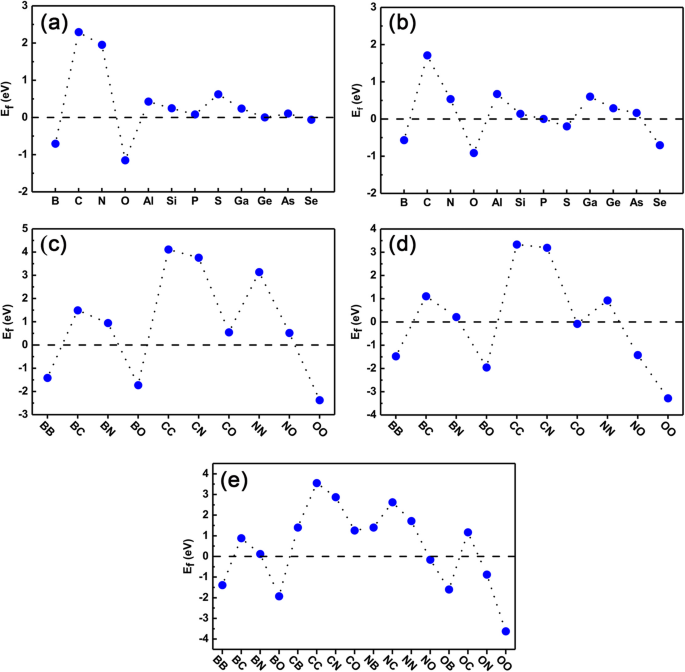
Energia de formação de todos os sistemas dopados. A energia de formação calculada de sistemas de dopagem e co-dopagem de elemento único. a , b são o átomo de Ge substituído por dopante e o átomo de P, respectivamente; c - e Os co-dopantes substituídos por dois átomos de Ge, dois átomos de P e um átomo de Ge e um átomo de P, respectivamente
Quanto à energia de formação de co-dopagem, Fig. 8c-e são as energias de formação de co-dopagem com onde dois dopantes ocupam as posições de dois átomos Ge (denotados como sítios Ge-Ge), dois átomos P (denotados como PP locais), um átomo Ge e um átomo P (denotados como locais Ge-P), respectivamente. Aqui, mostramos apenas os resultados dos dopantes de B, C, N e O como exemplo. Para locais Ge-Ge e locais P-P, a energia de formação de co-dopagem pode ser estimada aproximadamente pela média das energias de formação de dopagem de elemento único separadamente. Claramente, para co-dopagem BB, BO e OO em locais Ge-Ge e co-dopagem BB, BO, NO e OO em locais P-P, as energias de formação são relativamente pequenas e podem ser realizadas facilmente no experimento. No entanto, para co-dopagem CC, CN e NN em sítios Ge-Ge e co-dopagem CC e CN em sítios P-P, as energias de formação são relativamente maiores, indicando que são difíceis de dopar no experimento. Para a co-dopagem de sítios Ge-P, como mostrado na Fig. 8e, a energia de formação torna-se mais complexa do que a co-dopagem de sítios Ge-Ge ou P-P porque há transferência de carga entre os dopantes. De qualquer forma, o co-dopagem BB, BO e OO têm as energias de formação menores, enquanto o co-dopagem CC, CN e NN têm energias de formação maiores. Em geral, a energia de formação depende muito do número de elétrons de valência do dopante. Especificamente, quando os dois dopantes com um elétron a menos (mais) do que os átomos substituídos, a energia de formação do sistema co-dopado é menor (mais alta) do que a dos dopantes individuais correspondentes, como sítios Ge co-dopados BB (NN). Isso ocorre porque existe competição entre a energia diminuída (aumentada) de elétrons reduzidos (aumentados) de sistemas dopados e a repulsão de Coulomb. Para o co-dopagem buraco-buraco, a energia do primeiro caso é muito maior do que o último caso, resultando assim na energia de formação bastante diminuída em sistemas de co-dopagem como o BB, enquanto para o co-dopagem elétron-elétron, ambos os casos anteriores e posteriores levam à energia de formação mais alta, como NN. No entanto, para sistemas co-dopados com buraco e elétron, como sítios Ge co-dopados com BN, a energia de formação é dramaticamente mais baixa do que os casos com dopagem única correspondentes. Isso ocorre porque em tal sistema co-dopado, não há ganho de energia de elétrons adicionados ou reduzidos nos sistemas, e a interação de Coulomb desempenha um papel decisivo na formação de dopantes co-dopados. Ao todo, considerando nossos estudos anteriores de dopagem de elementos em fosforeno preto, deve-se ressaltar que nossos estudos atuais possuem um certo grau de universalidade e são esperados para a aplicação de outras monocamadas semicondutoras 2D, como BN, MoS 2 , e assim por diante.
Finalmente, para verificar a estabilidade dos sistemas dopados acima em comparação com o caso não dopado, realizamos o AIMD (dinâmica molecular ab initio) para mostrar a energia vs tempo, como mostrado na Fig. 9a-f. Podemos ver claramente que a amplitude oscilatória será convergente enquanto o tempo durar o suficiente (~ 4 ps), implicando que os sistemas dopados não entrarão em colapso contra a flutuação térmica de até 300 K para GeP dopado com C 3 na Fig. 9a. Mesmo para o GeP dopado com átomo C mais ativo 3 , a temperatura extrema pode ser de até 300 K, conforme mostrado na Fig. 9c. Além disso, também tomamos a substituição de metal Al no local Ge como um exemplo, o resultado calculado é mostrado na Fig. 9e e f, a partir da qual podemos ver claramente que a amplitude de oscilação de energia diminui gradualmente com o tempo de duração, o que significa que a energia poderia ser convergente desde que o tempo seja suficiente e a estrutura do sistema dopado seja termicamente estável contra oscilações térmicas. Portanto, podemos esperar que tais sistemas dopados possam ser realizados em experimentos posteriores, dado o GeP de alta qualidade 3 monocamada é preparada.
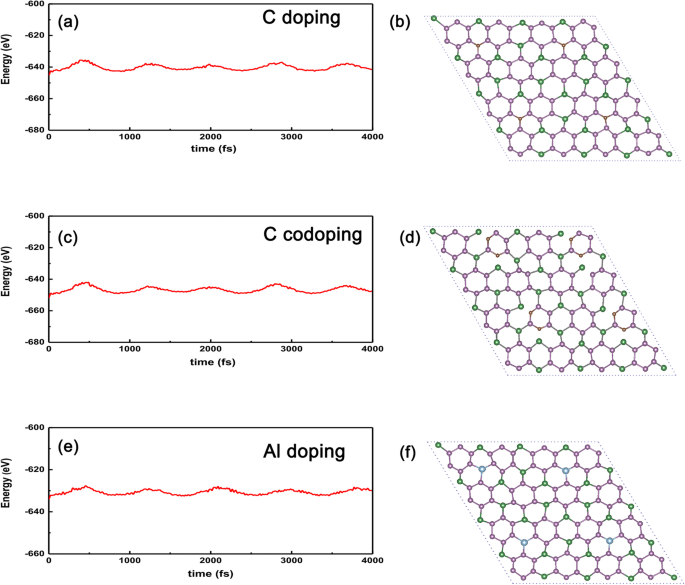
AIMD para os sistemas dopados com átomos C, dois C e Al. AIMD confirma a estabilidade térmica de a GeP dopado com átomo C 3 com substituição de átomo de Ge, c dois átomos de C dopados com GeP 3 com a substituição de dois átomos P, e e GeP dopado com átomo de Al 3 com substituição a 300 K. As estruturas em b C, d dois átomos C e f Al correspondem às suas estruturas finais após 4000 fs
No final, queremos discutir a confiabilidade do nosso estudo aqui apresentado. Nossas conclusões apresentadas aqui são resultados previstos teoricamente, mas são altamente confiáveis. Isso ocorre porque nosso material hospedeiro usado aqui foi relatado e sua fase em massa de GeP em camadas 3 já existe [26]. Portanto, nossos fenômenos relacionados induzidos por dopagem estudados precisam ser confirmados no experimento, uma vez que a monocamada GeP 3 é realizado ainda mais. Então, a dopagem dos átomos correspondentes pode ser conduzida. Para simplificar, dopando elétron ou buraco na monocamada GeP 3 poderia ser realizado pela adsorção de algumas moléculas.
Conclusão
Em resumo, investigamos as propriedades eletrônicas dos dopantes do grupo III a VI em 2D GeP 3 monocamada e descobrir que o dopado GeP 3 com substituição no local Ge exibe oscilações semicondutoras de metal em função do número de elétrons de valência dos dopantes, enquanto tais oscilações são revertidas com substituição no local P. Com base nos resultados de dopantes individuais, podemos propor as propriedades condutoras de co-dopagem em GeP 3 , que pode ser obtido por uma operação lógica simples. Finalmente, calculamos as energias de formação de vários dopantes e descobrimos que alguns dos sistemas co-dopados, especialmente para o co-dopagem elétron-buraco e buraco-buraco, são mais energeticamente favoráveis por causa da atração Coulomb. Nossos resultados não apenas apresentam um novo fenômeno, mas também propõem uma rota intrigante para ajustar as propriedades eletrônicas em semicondutores binários 2D.
Disponibilidade de dados e materiais
Os conjuntos de dados gerados durante e / ou analisados durante o estudo atual estão disponíveis junto ao autor correspondente mediante solicitação.
Abreviações
- 1D:
-
Unidimensional
- 2D:
-
Bidimensional
- AIMD:
-
Dinâmica molecular ab initio
- BP:
-
Fosforeno preto
- CBM:
-
Banda de condução mínima
- DFT:
-
Teoria da densidade funcional
- HSE06:
-
Densidade híbrida funcional
- PBE:
-
Perdew-Burke-Ernzerhof
- PDOS:
-
Densidade parcial de estados
- VBM:
-
Banda de valência máxima
Caracterização estrutural da cadeia auto-montada como a nanoestrutura do casco do Fe-FeOx Core
Progresso da pesquisa em nanoestruturas de óxido de manganita perovskita dopada com terras raras
Nanomateriais
- Valência e estrutura cristalina
- Elétrons e “buracos ''
- Remoção adsortiva de íons de cobre (II) de solução aquosa usando um nanoadsorvente de magnetita de resíduos de escala de moinho:Síntese, caracterização, adsorção e modelagem cinética Estud…
- Preparação de nanoesferas poliméricas impressas com íons de paládio (II) e sua remoção de paládio (II) da solução aquosa
- Síntese e atividade de oxidação de CO de 1D óxido binário misto CeO2-LaO x catalisadores de ouro suportados
- Nanomontagens de ácido 5-aminolevulínico-esqualeno para fotodetecção e terapia de tumor:estudos in vitro
- Síntese e caracterização de BiOCl modificado e sua aplicação na adsorção de corantes de baixa concentração de solução aquosa
- Morfologia, estrutura e propriedades ópticas de filmes semicondutores com Nanislands GeSiSn e camadas deformadas
- Otimização da absorção de banda larga e multibanda de grafeno monocamada em frequências ópticas de ressonâncias de dipolo magnético múltiplo em metamateriais
- Monocamada de g-GaN adsorvida por metal alcalino:funções de trabalho ultrabaixo e propriedades ópticas