


Conversa do antiferromagnético MnBr2 para a monocamada ferromagnética Mn3Br8 com MAE grande
Resumo
Uma necessidade premente na spintrônica de baixa energia são os ferromagnetos bidimensionais (2D) com temperatura de Curie acima da temperatura do nitrogênio líquido (77 K) e anisotropia magnética considerável. Estudamos Mn 3 Br 8 monocamada que é obtida através da indução de vacância de Mn em 1/4 da população em MnBr 2 monocamada. Essa configuração defeituosa é projetada para alterar a estrutura de coordenação do Mn-d 5 e alcançar o ferromagnetismo com energia de anisotropia magnética considerável (MAE). Nossos cálculos mostram que Mn 3 Br 8 monocamada é um meio-metal ferromagnético (FM) com temperatura de Curie de 130 K, grande MAE de - 2,33 meV por unidade de fórmula e momento magnético atômico de 13 / 3μ B para o átomo Mn . Além disso, Mn 3 Br 8 a monocamada mantém-se FM sob pequena cepa biaxial, cuja temperatura de Curie abaixo de 5% da deformação compressiva é de 160 K. Além disso, tanto a cepa biaxial quanto o dopagem de portadores aumentam o MAE, o que contribui principalmente pela energia de anisotropia magneto-cristalina (MCE). Nossa estrutura defeituosa projetada de MnBr 2 monocamada fornece uma maneira simples, mas eficaz de obter ferromagnetismo com grande MAE em materiais 2D.
Introdução
Spintrônica, explorando o spin do elétron e o momento magnético associado, tem atraído grande atenção durante as últimas décadas [1], por causa de suas vantagens exclusivas sobre dispositivos baseados em carga. A recente realização de ferromagnetos bidimensionais (2D) com ordenação magnética de longo alcance em temperatura finita [2, 3] é de grande importância para a spintrônica em nanoescala e aplicações relacionadas e inspira esforços tremendos em investigações e fabricação de ferromagnetos 2D [4,5 , 6,7,8,9].
Os primeiros dois ferromagnetos 2D com espessura atômica foram alcançados em 2017, ou seja, monocamada CrI 3 [2] e bicamada Cr 2 Ge 2 Te 6 [3]. Infelizmente, ambas as temperaturas Curie são mais baixas do que a temperatura do nitrogênio líquido (77 K), o que limita suas aplicações realistas. Além da temperatura de Curie, a anisotropia magnética considerável e o momento magnético também são indispensáveis para a aplicação prática. Grande energia de anisotropia magnética (MAE) implica no benefício do ordenamento magnético contra a flutuação de calor, e a possibilidade de redução do tamanho de grão por bit de informação; MAE pequeno pode resultar em super-paramagnético em vez de ferromagnético. Grande momento magnético fornece maior sensibilidade, maior eficiência e maior densidade para spintrônica. Elementos pesados são mais propensos a trazer grande MAE devido ao seu forte efeito spin-orbital coupling (SOC) [10]. Uma série de materiais 2D FM compostos de elementos pesados foram previstos com grande MAE, como CrI 3 [11], CrAs [12], CrSeI [13], CrSiTe 3 [14], CrWI 6 [15], FeBr 2 e FeI 2 monocamadas [16]. Além disso, o momento magnético local no átomo de Mn de MXenes Mn 2 NF 2 e Mn 2 N (OH) 2 é 4,5 μ B por átomo de Mn [17], que é o maior entre os materiais FM 2D relatados.
Desde CrI 3 monocamada foi sintetizada com sucesso, haletos de metais de transição têm atraído muitas atenções [18,19,20,21,22,23,24,25,26,27]. O efeito Spin Seeback foi observado na bicamada MnF 2 [20]; algumas camadas de CrI 3 foi implementado nas junções de tunelamento magnético (MTJ) [21]; NiCl 3 foi previsto que a monocamada seja um novo semicondutor Dirac spin-gapless (SGS) [22]. Particularmente, MnBr 2 a monocamada é antiferromagnética com 0,25 meV MAE ao longo da direção perpendicular ao plano com base nos cálculos dos primeiros princípios [16]; Mn 2+ os íons estão no d 5 estado de alta rotação com momento magnético de 5μ B [16, 26]. Esses resultados implicam nos potenciais de MnBr 2 como ferromagneto monocamada com grande momento magnético. O principal problema é como converter o acoplamento AFM entre os íons Mn em acoplamento FM.
Densidade significativa de vacância de Mn foi observada experimentalmente em LaMnO 3 filmes finos [28] e a concentração de defeitos podem ser controlados regulando o processo de síntese deliberadamente por meio de irradiação de partículas de alta energia ou corrosão química [29]. Neste contexto, projetamos o Mn 3 Br 8 monocamada induzindo vaga de Mn única para MnBr 2 monocamada. A vacância muda a estrutura de coordenação do átomo Mn e quebra o d 5 configuração, que pode converter o acoplamento antiferromagnético em acoplamento ferromagnético e trazer grande MAE devido ao átomo de Br pesado. Como esperamos, Mn 3 Br 8 monocamada é FM e tem grande MAE de - 2,33 meV por unidade de fórmula, o momento magnético para cada átomo de Mn é 13 / 3μ B . Considerando a fácil introdução de deformação via flexão de substratos flexíveis [30,31,32,33], alongamento do substrato elástico [33,34,35], explorando a incompatibilidade de expansão térmica [33, 36], e assim por diante [33], e o controle efetivo da polarização de spin via dopagem eletrostática [37, 38], também estudamos o Mn 3 Br 8 monocamada sob cepa biaxial e dopagem de portadores. Nossos resultados mostram que Mn 3 Br 8 a monocamada mantém-se FM com temperatura de Curie aumentando sob pequena tensão biaxial. Além disso, tanto a deformação biaxial quanto o doping de portador podem aumentar o MAE.
Métodos computacionais
Todos os cálculos no presente estudo foram realizados através da adoção do método da teoria da função de densidade polarizada de spin (DFT), conforme implementado no ab-initio de Viena pacote de simulação (VASP) [39]. As interações entre elétrons e núcleos foram descritas pelo método de onda aumentada do projetor (PAW) [40, 41], e as interações de correlação de troca eletrônica foram descritas por Perdew-Burke-Ernzerhof (PBE) funcional dentro da aproximação de gradiente generalizado (GGA) método [42]. Os termos U de Hubbard foram adotados para calcular a interação fortemente correlacionada [43]; um parâmetro de interação coulombiana efetiva no local (U) de 4 eV e uma energia de troca (J) de 1 eV, que foi adotada para estudar materiais 2D incorporados por Mn, foram usados para os elétrons Mn-d [44]. A integração da zona de Brillouin foi realizada adotando-se a malha 9 × 9 × 1 k com base no esquema Monkhorst-Pack [45]. Os espectros de fônons foram calculados usando o código Phonopy [46], que é implementado no pacote VASP. Um espaço de vácuo de 20 Å foi adicionado ao longo da direção perpendicular à superfície da monocamada para evitar a interação entre as camadas adjacentes. A energia de corte para o conjunto de base da onda plana foi definida como 500 eV. O critério de convergência para a energia e força total foi definido como 1 × 10 –6 eV e 0,01 eV / Å, respectivamente.
Resultados e discussões
Energia de clivagem, estado fundamental e estabilidade do MnBr 2 monocamada
As constantes de rede otimizadas de MnBr em massa 2 são a =b =3,95 Å, consistentes com o resultado experimental anterior ( a = b =3,87 Å) [25]. Em primeiro lugar, exploramos a viabilidade de esfoliar MnBr 2 monocamada do MnBr em massa 2 . A Figura 1a apresenta o método conhecido, eficaz e amplamente aprovado de cálculo da energia de clivagem [47,48,49]. Especificamente, a energia de clivagem foi obtida calculando a variação da energia total do estado fundamental em relação à distância de separação \ (d \) entre as duas partes da fratura como mostrado na Fig. 1b, as constantes de rede de a e b são fixados como os valores no estado de equilíbrio da massa MnBr 2 . As interações vdW de longo alcance entre camadas foram descritas pelo esquema DFT-D2 de Grimme [50, 51]. A energia total aumenta com a distância de separação e então converge lentamente como mostrado na Fig. 1b. A energia de clivagem calculada é 0,10 J / m 2 , que é menor em comparação com a energia de clivagem entre as duas partes da fratura de grafite (0,35 J / m 2 ) [52], demonstrando a viabilidade de obtenção de MnBr 2 monocamada através do método de esfoliação micro-mecânica.
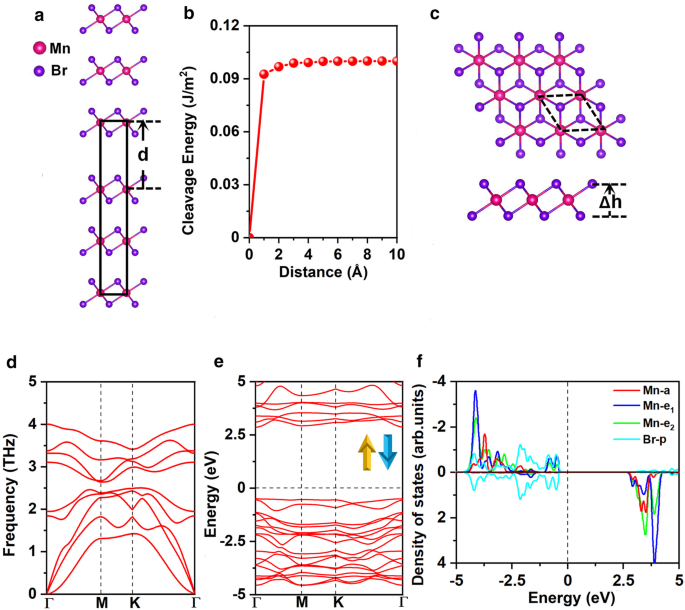
a Modelo em massa de MnBr 2 usado para calcular a energia de clivagem e b a energia de clivagem como uma função da distância de separação \ (d \) entre duas partes fraturadas (a distância intercamada de equilíbrio é definida como 0). c Vistas superior e lateral, d espectro de fônons, e estrutura de banda eletrônica para canais de spin e f densidade projetada de estados (PDOS) de orbitais Mn-d e orbitais Br-p para MnBr 2 monocamada. Δ h representa a distância vertical entre dois planos de haletos. A célula primitiva é distribuída em linhas tracejadas pretas. O nível de Fermi para estrutura de banda e DOS é definido como 0 eV
MnBr 2 monocamada tem a simetria \ (C _ {{{3} v}} \) como mostrado na Fig. 1c; cada átomo de Mn é cercado por 6 átomos de Br vizinhos, formando um octaédrico [MnBr 6 ] 4− unidade. Como mostrado na Fig. 2a eb, três configurações magnéticas possíveis, a saber, estados não magnéticos (NM), ferromagnéticos (FM) e antiferromagnéticos (AFM) são considerados. Os estados de spin alto e baixo do íon Mn são considerados. Nossos resultados mostram que os íons Mn do estado FM estão em spin baixo com d 1 configuração, enquanto os íons Mn no estado AFM estão em alta rotação com d 5 configuração. O estado fundamental de MnBr 2 monocamada é o estado AFM, que é mais estável do que os estados NM e FM em 3,91 eV e 0,72 eV por unidade de fórmula, respectivamente (Arquivo adicional 1:Tabela. S1). O MAE é de 0,25 meV, o valor positivo indicando que o eixo de fácil magnetização está ao longo das direções fora do plano, concordando com o resultado anterior [16]. As constantes de rede otimizadas são a = b =3,95 Å, o mesmo com as constantes de rede da massa MnBr 2 . O comprimento da ligação Mn-Br é 2,73 Å, e a distância vertical entre os dois planos de haleto é 3,03 Å.
Diagramas esquemáticos para a ferromagnético e b configurações antiferromagnéticas para MnBr 2 monocamada
A estabilidade do MnBr 2 a monocamada foi investigada com o cálculo da energia de formação, espectro de fônons e constantes elásticas. A energia de formação é calculada como:
$$ E _ {{{\ text {form}}}} =E _ {{{\ text {MnBr}} _ {{2}}}} - E _ {{{\ text {Mn}}}} - 2E _ {{ {\ text {Br}}}} $$
onde \ (E _ {{{\ text {MnBr}} _ {{2}}}} \) representa a energia de MnBr 2 monocamada, \ (E _ {{{\ text {Mn}}}} \) e \ (E _ {{{\ text {Br}}}} \) são as energias dos átomos de Mn e Br em suas estruturas em massa, respectivamente. O \ (E _ {{{\ text {form}}}} \) calculado é - 1,87 eV por átomo; o valor negativo significa que a formação é exotérmica e MnBr 2 monocamada é energeticamente favorável. Além disso, nosso espectro de fônon calculado (Fig. 1d) para MnBr 2 monocamada não mostra frequência negativa em toda a zona de Brillouin, indicando dinamicamente estável. Além disso, as constantes elásticas calculadas (Arquivo adicional 1:Tabela S2) obedecem aos critérios de Born-Huang [53] de \ (C_ {11}> 0 \), \ (C_ {11} C_ {22} - C_ {12 } ^ {2}> 0 \) e \ (C_ {66}> 0 \), confirmando que MnBr 2 monocamada é mecanicamente estável. A rigidez no plano calculada é 26,98 J / m 2 , cerca de 75% do MnPSe 3 (36 J / m 2 ) [49], e 15% do MoS 2 monocamada (180 J / m 2 ) [54]. Além disso, MnBr 2 monocamada demonstra maior flexibilidade e a capacidade de sustentar maior tensão de tração em comparação com MoS 2 monocamada (11%) [54]. Isso pode ser atribuído a ligações iônicas para MnBr 2 monocamada contra as ligações covalentes de MoS 2 monocamada. A análise da deformação relacionada às constantes elásticas indica que ela pode suportar seu peso (ver detalhes no SI).
A estrutura da banda eletrônica de MnBr 2 monocamada é mostrada na Fig. 1e, indica que MnBr 2 monocamada é um semicondutor com um gap direto de 3,35 eV. A banda de valência máxima (VBM) e a banda de condução mínima (CBM) estão localizadas no ponto \ (\ Gamma \). Para obter uma visão das estruturas eletrônicas, a densidade projetada de estados (DOS) para os orbitais Mn-d e Br-p são apresentados na Fig. 1f. Os cinco orbitais d do íon Mn dividem-se em \ (a (d _ {{z ^ {2}}}) \), \ (e_ {1} (d_ {xz} + d_ {yz}) \), e \ ( e_ {2} (d_ {xy} + d _ {{x ^ {2} - y ^ {2}}}) \) grupos de acordo com a simetria \ (C _ {{{3} v}} \). A pior análise de carga sugere que cada átomo de Mn doa dois elétrons para os dois átomos de Br vizinhos. Assim, os cinco orbitais d em um canal de spin são totalmente ocupados pelos cinco elétrons d do Mn 2+ íons. Correspondentemente, os dois Mn 2+ íons na célula unitária estão no d 5 estado de alta rotação com o momento magnético de 5μ B / - 5μ B , o Br 1− os íons estão no estado de baixa rotação de 4p 6 com momento magnético desprezível de - 0,02μ B (Arquivo adicional 1:Fig. S1 (a)). De acordo com a regra de Goodenough – Kanamori – Anderson (GKA), tal configuração sempre fornece acoplamento antiferromagnético [55].
Estabilidade, propriedades eletrônicas e magnéticas de Mn 3 Br 8 monocamada
A vaga Mn foi introduzida para quebrar o d 5 configuração do Mn 2+ íons. A vaga única de Mn é introduzida na supercélula \ (2 \ vezes 2 \ vezes 1 \) de MnBr 2 monocamada, que fornece o Mn 3 Br 8 monocamada. Como mostrado na Fig. 3a, cada átomo de Mn tem quatro átomos de Mn vizinhos mais próximos e se liga a seis átomos de Br, formando um octaédrico distorcido [MnBr 6 ] unidade. Cinco estados magnéticos (NM, FM, FIM, AFM-1 e AFM-2) mostrados na Fig. 4 foram considerados. Nossos resultados indicam que o estado FM é o estado fundamental, que é mais estável do que os outros quatro por 9,84 eV, 32,90 meV, 129,85 meV e 97,65 meV por unidade de fórmula, respectivamente. A constante de rede otimizada ainda é 3,95 Å. Diferente de MnBr 2 monocamada, Mn 3 Br 8 monocamada tem 2 tipos de ligações Mn-Br (Fig. 3b). As ligações entre o átomo Mn e os dois átomos Br centrais (\ (d _ {{\ text {Mn-Br1,2}}} \)) são 2,76 Å, enquanto as outras ligações Mn-Br (\ (d _ {{\ text {Mn-Br3,4,5,6}}} \)) são 2,59 Å. A distância vertical entre os dois planos de haletos é 3,33 Å.
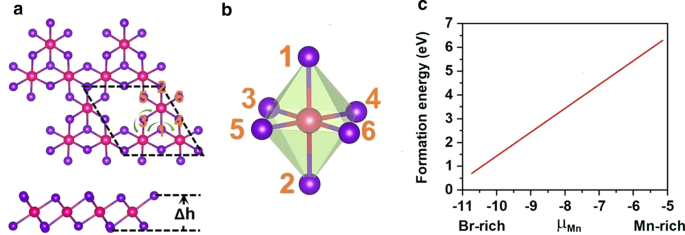
a Vistas superior e lateral do Mn 3 Br 8 monocamada, \ (\ Delta h \) representa a distância vertical entre dois planos de haletos. A célula primitiva circula em linhas tracejadas pretas; as linhas da seta verde mostram dois caminhos diferentes da interação de supertroca. b Estrutura do MnBr distorcido 6 octaedro. c Energias de formação para vacância única de Mn em função do potencial químico de Mn (μMn)
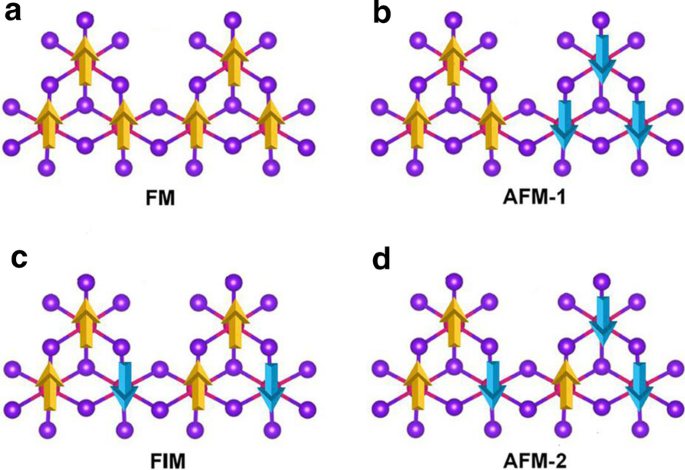
Diagramas esquemáticos para a ferromagnético, b antiferromagnético-1, c ferrimagnético e d configurações antiferromagnéticas-2 para Mn 3 Br 8 monocamada
Para verificar a viabilidade de induzir a vacância de Mn, primeiro calculamos as energias de formação de vacância em ambientes ricos em Mn e Br através das seguintes equações,
$$ E _ {{F ({\ text {Mn-rico}})}} {\ text {=}} E _ {{{\ text {Mn}} _ {3} {\ text {Br}} _ {8 }}} - (4 \ vezes E _ {{{\ text {MnBr}} _ {{\ text {2}}}}} - \ mu _ {{{\ text {Mn-max}}}}) $$ $$ E _ {{F {\ text {(Br-rico)}}}} {=} E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8} }}} - (4 \ vezes E _ {{{\ text {MnBr}} _ {{2}}}} - \ mu _ {{\ text {Mn-min}}}) $$
onde \ (E _ {{{\ text {Mn}} _ {{3}} {\ text {Br}} _ {{8}}}} \) e \ (E _ {{{\ text {MnBr}} _ {{2}}}} \) representam as energias totais do Mn 3 Br 8 e MnBr 2 monocamadas, \ (\ mu _ {{\ text {Mn-max}}} \) é o potencial químico de Mn sob um ambiente rico em Mn, que é calculado como a energia do átomo de Mn em sua estrutura em massa, \ (\ mu_ { {\ text {Mn-min}}} \) é o potencial químico de Mn sob o ambiente rico em Br, que é calculado como:
$$ \ mu _ {{\ text {Mn-min}}} =E _ {{{\ text {MnBr}} _ {{2}}}} - 2 \ vezes \ mu _ {{\ text {Br-max}} } $$
onde \ (\ mu _ {{\ text {Br-max}}} \) é o potencial químico de Br e calculado como a energia do átomo de Br na fase gasosa. Como mostrado na Fig. 3c, as energias de formação sob ambiente rico em Mn / rico em Br são 6,30 / 0,71 eV por vacância em Mn, indicando que a formação de vacância em Mn é energeticamente mais favorável sob o ambiente rico em Br. Na verdade, a vaga S foi experimentalmente alcançada no MoS 2 monocamada [56], e a energia de formação prevista da vacância de S sob o ambiente rico em S é de 2,35 eV [57]. Além disso, estruturar nanoarquitetura porosa como β-FeOOH / PNGNs (redes de grafeno dopado com nitrogênio poroso) pode induzir vacância de Fe significativa [58], e o método de Bridgman foi adotado para induzir vacância de Fe de ordenação. Esperamos também que esses métodos sejam aplicáveis para induzir a vacância de Mn [59]. Além disso, não há frequência negativa encontrada no espectro de fônons de Mn 3 Br 8 monocamada mostrada na Fig. 5a, comprovando a estabilidade dinâmica. Esses resultados aprovam nosso projeto de introdução da vacância de Mn para introduzir o ferromagnetismo.
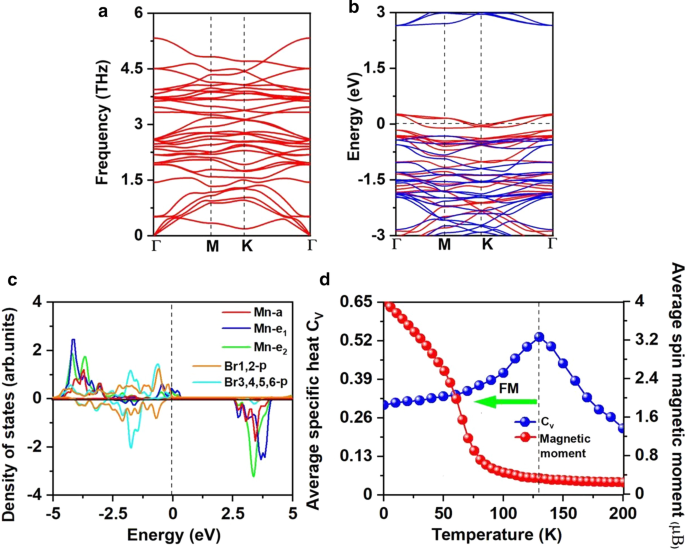
a Espectros do fonema, b estrutura de banda eletrônica com resolução por spin e c densidade projetada de estados (PDOS) de orbitais Mn-d e orbitais Br-p para Mn 3 Br 8 monocamada. d Momentos magnéticos no local de átomos de Mn e o calor específico C v em função da temperatura com base no modelo de Heisenberg para Mn 3 Br 8 monocamada. O nível de Fermi para estrutura de banda e PDOS é definido como 0 eV
O ferromagnetismo de Mn 3 Br 8 atributos de monocamada para a interação de supertroca FM. De acordo com a regra de Goodenough-Kanamori-Anderson (GKA) [55], a interação de supertroca entre os íons Mn é FM quando o ângulo Mn-Br-Mn está em torno de 90 °. Nessa configuração (arquivo adicional 1:Fig. S2), o orbital Mn-d tende a acoplar AFM com orbitais Br-p ortogonais diferentes e, portanto, espera-se que o acoplamento magnético indireto Mn-Mn seja FM. Mas se cada íon Mn tiver 5 elétrons desemparelhados como MnBr 2 monocamada, a supertroca é AFM, embora o ângulo Mn-Br-Mn seja próximo a 90 ° porque não há orbitais Mn-d de spin-up vazios restantes em MnBr 2 os elétrons d de monocamada e spin-up não podem saltar entre o local Mn vizinho [60]. Existem dois caminhos diferentes de interação de supertroca no Mn 3 Br 8 (Fig. 3a), e ambos são FM. Um envolve átomos Br1,2 centrais com comprimentos de ligação Mn-Br de 2,76 Å e ângulos Mn-Br-Mn de 87,5 °; o outro envolve átomos Br3,4,5,6 com comprimento de ligação Mn-Br de 2,59 Å e ângulos Mn-Br-Mn de 95 °. As interações hibridizadas entre orbitais p de átomos Br3,4,5,6 e orbitais Mn-d são mais fortes do que a hibridização p-d envolvendo átomos Br1,2, como mostrado na Fig. 5c, particularmente de - 2 eV a - 1,4 eV. Enquanto de 1,4 a - 0,9 eV, o p - d hibridização envolvendo átomos Br1,2 são dominados.
A pior análise de carga sugere que cada átomo de Mn doa 8/3 elétrons para os átomos de Br vizinhos. Assim, os íons Mn estão no Mn 8/3 + Estado. Conforme mostrado na Fig. 5c, os 13/3 elétrons de cada íon Mn preenchem o canal de spin-up do orbital d, enquanto o Br 1− os íons estão no estado de baixa rotação de 4p 6 . Assim, o momento magnético de cada Mn 8/3 + íon é 13 / 3μ B ; o momento magnético de Br 1− os íons são desprezíveis (Arquivo adicional 1:Fig. S1 (b)). Indução de ferromagnetismo por vacância também pode ser observada para o d 0 sistemas, como ZnS e ZnO [61, 62], a vacância única pode induzir um momento magnético tão grande quanto 2μ B [61] . Para cada íon Mn, 2/3 d-orbital está desocupado; o canal de spin-up de ambos os orbitais \ (e_ {1} \) e \ (e _ {{2}} \) está parcialmente ocupado e cruzando o nível de Fermi, resultando em semimetalicidade. O caráter semimetálico também pode ser observado a partir da estrutura de banda eletrônica com spin resolvido mostrada na Fig. 5b. O canal de spin-up é metálico, enquanto o canal de spin-down é semicondutor com o gap indireto de 2,97 eV; o VBM / CBM localiza-se no ponto \ ({\ text {M}} \) / \ (\ Gamma \). O valor do gap é próximo ao MnP (2,86 eV) [63], MnAs (2,92 eV) [63] e Ni 2 NÃO 2 (2.98 eV) [64], que é grande o suficiente para evitar o spin-flip termicamente excitado. Comparando com o MnBr 2 monocamada, tanto o VBM quanto o CBM do canal semicondutor ficam mais próximos do nível de Fermi. O CBM ainda é dominado pelos átomos de Mn, enquanto o VBM é dominado pelos novos átomos Br1,2. Enquanto isso, o canal semicondutor converte de direto em indireto e o gap diminui. O fenômeno semelhante foi observado em MnCl 2 monocamada com funcionalização H [60].
As direções de magnetização são determinadas pela energia de anisotropia magnética (MAE). O MAE de sólidos surge de dois contribuintes, a saber, a energia magneto-cristalina (MCE) relacionada ao acoplamento spin-órbita (SOC) e a energia de anisotropia dipolar magnética (MDE) atribuída pela interação dipolo-dipolo magnetoestático. O MDE nos materiais isotrópicos 3D, como bcc Fe e fcc Ni, é muito pequeno. Mas para materiais de baixa dimensão compostos de átomos de metal de transição com grande momento magnético, o MDE não deve ser ignorado [65,66,67]. O MCE é definido como a diferença entre a energia de magnetização ao longo das direções no plano (100 ou 010) e fora do plano (001), levando-se em consideração o SOC. O MDE é obtido como a diferença de \ (E_ {d} \) entre as magnetizações no plano e fora do plano. \ (E_ {d} \) em unidades atômicas de Rydberg é dado por [65, 66]
$$ E_ {d} =\ sum \ limits_ {ij} {\ frac {{2m_ {i} m_ {j}}} {{c ^ {2}}}} M_ {ij} $$
onde a velocidade da luz, \ (c =274,072 \), \ (i / j \) são os vetores de posição atômica na célula unitária, e \ ({m} _ {i} / {m} _ {j} \ ) é o momento magnético atômico (μ B ) no local \ (i / j \). A constante dipolar magnética de Madelung \ (M_ {ij} \) é calculada via
$$ M_ {ij} =\ sum \ limits_ {R} {\ frac {1} {{\ left | {R + i + j} \ direita | ^ {3}}}} \ esquerda \ {{1 - 3 \ esquerda. {\ frac {{\ left [{(R + i + j) \ cdot \ mathop {m_ {i}} \ limits ^ {\ wedge}} \ right] ^ {2}}} {{\ left | {R + i + j} \ right | ^ {2}}}} \ right \}} \ right. $$
onde \ (R \) são os vetores da rede. Em um material 2D, uma vez que todos os \ (R \) e \ (i \) estão no plano, o segundo termo seria zero para a magnetização fora do plano, resultando no positivo \ (M_ {ij} \ ), enquanto \ (M_ {ij} \) é negativo para uma magnetização no plano [67]. Portanto, o MDE se refere ao momento magnético do metal de transição e sempre prefere a magnetização no plano.
O MCE calculado para Mn 3 Br 8 monocamada é - 1,90 meV por unidade de fórmula (Fig. 6a), muito maior do que aquelas de Fe em massa (0,001 meV por átomo) e Ni (0,003 meV por átomo) [68], e maior do que a da monocamada de Fe em Rh (111) (0,08 meV por átomo) [69], sugerindo que a magnetização do Mn 3 Br 8 monocamada é termicamente estável. A relação entre o MCE e o ângulo azimutal pode ser descrita pela seguinte equação [70]:
$$ {\ text {MCE}} (\ theta) =A \ cos ^ {2} (\ theta) + B \ cos ^ {4} (\ theta) $$
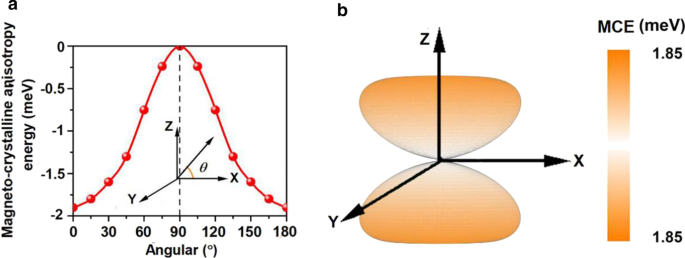
Variação da energia de anisotropia magneto-cristalina (MCE) a com respeito ao ângulo azimutal e b no espaço para Mn 3 Br 8 monocamada
onde \ (A \) e \ (B \) são as constantes de anisotropia e \ (\ theta \) é o ângulo azimutal. O resultado do ajuste é mostrado no Arquivo Adicional 1:Figs. S3. Além disso, a evolução do MCE com o eixo de rotação girando por todo o espaço é ilustrada na Fig. 6b. O MCE dentro do plano xy não mostra diferença, mas atinge o valor máximo ao longo da direção perpendicular ao plano xy, confirmando a forte anisotropia magnética. O MDE é - 0,43 meV por unidade de fórmula, e MAE (MCE + MDE) é - 2,33 meV por unidade de fórmula. O valor negativo indica que o eixo de magnetização fácil está ao longo das direções no plano. O MDE não muda a direção da magnetização, mas a aprimora. Além disso, o MAE de Mn 3 Br 8 monocamada é muito maior do que MnBr 2 monocamada, provando mais uma vez a eficácia do nosso design.
Além disso, calculamos o \ (T_ {c} \) para FM Mn 3 Br 8 monocamada através da realização de simulações de Monte Carlo (MC) com base no modelo de Heisenberg, que tem se mostrado o método eficaz para prever \ (T_ {c} \) para materiais 2D [11, 15, 48, 58, 71,72 , 73,74,75,76]. Nossa estimativa de \ (T_ {c} \) de CrI 3 monocamada é de 42 K (arquivo adicional 1:Fig. S4) [76], concordando bem com o valor medido experimental [2] e resultados de cálculos anteriores [15, 58, 71, 72, 74, 76], o que prova a precisão de nosso método adotado. O spin-hamiltoniano incluindo a interação magnética da vizinhança mais próxima (NN) é descrito como
$$ H =- \ soma \ limites_ {i, j} {JM_ {i} M_ {j}} $$
onde \ (J \) é o parâmetro de troca magnética NN, \ (M_ {i / j} \) é o momento magnético de íons Mn e integral próximo ao número de elétrons polarizados de spin com base no método de Monte Carlo [71, 77 , 78], \ (i \) e \ (j \) representam o par NN de íons Mn. O parâmetro de acoplamento magnético \ (J \) é calculado através da diferença de energia entre os estados FM e AFM como
$$ J {=} \ frac {{E _ {{{\ text {AFM1}}}} - E _ {{{\ text {FM}}}}}} {{16M ^ {2}}} $$
O \ (J \) calculado de íons NN Mn é 1,01 meV; o valor positivo indica a preferência de acoplamento FM.
O \ (J \) calculado dos íons NN Mn e a supercélula \ (100 \ vezes 100 \ vezes 1 \) contendo 20.000 vetores de momento magnético foram adotados para realizar as simulações de MC. As simulações em cada temperatura duram 10 5 degraus. Cada vetor de momento magnético gira aleatoriamente em todas as direções. A Figura 5d mostra a evolução do calor específico definido como \ (C _ {{_ {V}}} ={{\ left ({\ left \ langle {E ^ {2}} \ right \ rangle - \ left \ langle E \ direita \ rangle ^ {2}} \ direita)} \ mathord {\ left / {\ vphantom {{\ left ({\ left \ langle {E ^ {2}} \ right \ rangle - \ left \ langle E \ right \ rangle ^ {2}} \ right)} {K_ {B} T ^ {2}}}} \ right. \ kern- \ nulldelimiterspace} {K_ {B} T ^ {2}}} \) com temperatura, do qual obtivemos o \ (T_ {c} \) de 130 K para Mn 3 Br 8 monocamada localizando a posição do pico de \ (C_ {v} \), mais alta do que a temperatura do nitrogênio líquido (77 K), e \ (T_ {c} \) de CrI 3 (45 K) [2] e Cr 2 Ge 2 Te 6 (28 K) [3], CrX 3 (X =F, Cl, Br) (36 ~ 51 K) [11], CrXTe 3 (X =Si, Ge) (35,7 K, 57,2 K) [48]. Nossos cálculos demonstram que o FM Mn 3 Br 8 monocamada tem grande MAE e temperatura de Curie mais alta do que a temperatura do nitrogênio líquido.
Mn 3 Br 8 monocamada sob cepa biaxial e dopagem de portador
A engenharia de deformação tem se mostrado aplicável para muitos materiais 2D e eficaz para alterar os parâmetros estruturais, como comprimentos e ângulos de ligação, e ajustar as propriedades eletrônicas e magnéticas. Neste contexto, investigamos Mn 3 Br 8 monocamada sob a cepa biaxial variando de -5% a 5%. Acontece que Mn 3 Br 8 monocamada sob deformação biaxial de -5 a 5% mantém-se FM e o momento magnético atômico dificilmente muda. Conforme mostrado nas Figs. 7a e c, os ângulos entre dois átomos de Mn e átomos de Br1,2 (θ Mn-Br1,2-Mn ) são 84 ° -90 °, o que aumenta conforme a deformação e se aproxima gradualmente de 90 °. Os ângulos Mn-Br-Mn envolvendo átomos Br3,4,5,6 (θ Mn-Br3,4,5,6-Mn ) desviam gradualmente de 90 °, variando de 90 ° a 100 °. Assim, as interações de supertroca entre os íons Mn mediadas por diferentes orbitais Br-p ortogonais ainda são FM.
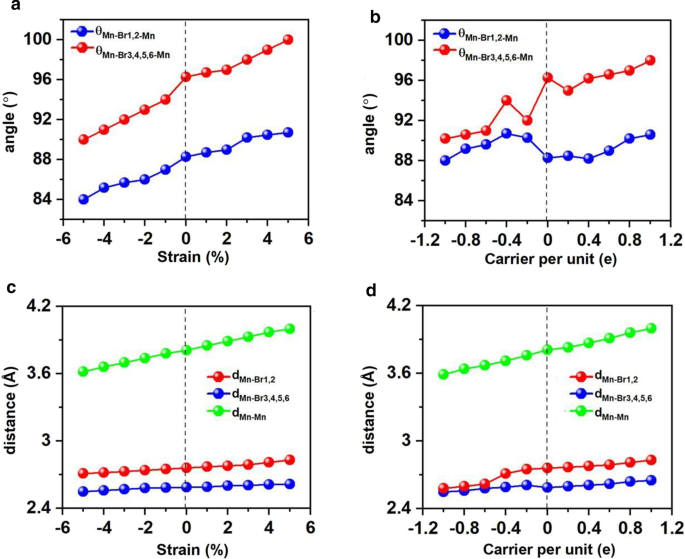
Variações de ângulos entre dois átomos de Mn e Br, a distância entre os átomos de Mn e Br e a distância entre os átomos de Mn vizinhos mais próximos em relação à cepa biaxial aplicada e ao dopagem de portador. Variação de a ângulo e c distância em relação à deformação biaxial, variações de b ângulo e d distância em relação ao doping de portador. Os valores positivos e negativos de dopagem de portador representam o dopagem de elétron e buraco, respectivamente
Ambas as distâncias Mn – Mn e Mn-Br aumentam monotonicamente à medida que a deformação muda de –5% a 5%. Correspondentemente, o parâmetro de troca sob a deformação biaxial apresentado na Fig. 8a diminui com a variação da deformação biaxial de –5% a 5% e atinge o maior valor (1,18 meV) sob a deformação biaxial de –5%. A temperatura Curie de Mn 3 Br 8 monocamada sob -5% de deformação biaxial é 160 K (Fig. 9a). Particularmente, as ligações Mn-Br sob a deformação de tração crescente tornam-se mais longas e os ângulos de Mn-Br3,4,5,6-Mn desviam de 90 °, que são a principal razão pela qual a interação de supertroca FM torna-se mais fraca. Consequentemente, a temperatura Curie diminui. É semelhante ao CrPTe 3 e FePS 3 monolayers [79]. Additionally, the MDE decreases with the increasing strain (Additional file 1:Fig. S5(b)); the MAE under –1% biaxial strain is the largest (–3.04 meV). The –5–5% strain does not cause large structural deformation for Mn3 Br8 monolayer, and the morphology of its band structures hardly changes. Mn3 Br8 monolayer keeps to be half-metallic. Both VBM and CBM in the semiconducting spin-channel move upward slightly to the higher energy as shown in Figs. 8c and 10; the band gap increases slowly with the increasing biaxial strain to 3.12 eV under 5% biaxial strain.

Variations of a the exchange parameter and b magnetic anisotropy energy (MAE) for Mn3 Br8 monolayer with respect to the applied biaxial strain and carrier doping. The variations of valence band maximum (VBM), conduction band minimum (CBM), and band gap in the semiconducting channel for Mn3 Br8 monolayer with respect to c the applied biaxial strain and d carrier doping ranging. Positive and negative values of the carrier doping represent the electron and hole doping, respectively

On-site magnetic moments of Mn atoms and the specific heat C v as function of temperature based on Heisenberg model for Mn3 Br8 monolayer a under -5% biaxial strain, with b 0.2e, c -0.6e, and d -0.8e carrier doping per formula unit. Positive and negative values represent the electron and hole doping, respectively

a - j Spin-resolved band structure for Mn3 Br8 monolayer under biaxial strain from -5% to 5%. The green arrow denotes the indirect band gap
Electron/hole doping always leads to VBM/CBM moving away from the Fermi level. Our calculations show that Mn3 Br8 monolayer with –1–1e (~ \(1.7 \times 10^{14} {\text{cm}}^{{ - 2}}\)) carrier doping per formula unit is still FM; the atomic magnetic moment of each Mn ion is still 13/3μB. As shown in Fig. 7b and d, with carrier doping from –1e to 1e per formula unit, the Mn-Br-Mn angles involving Br3,4,5,6 atoms are about 90° ~ 98°; the Mn-Br1,2-Mn angles are about 88° ~ 90°. The Mn–Mn and Mn-Br1,2 distances increase with the increasing electron doping. Mn3 Br8 monolayer with 0.2e and 0.4e carrier doping has larger magnetic exchange parameter (Fig. 8a). The Curie temperature at 0.2e electron doping is largest of 140 K (Fig. 9b). Additionally, with –1e ~ 0.2e doping, the MAE is along in-plane directions; the MDE decreases with the increasing electron doping. Under 0.4e doping, the MCE turns to be positive with the value of 0.41 meV per formula unit; the MAE is only 0.01 meV per formula unit with taking the MDE into account (Additional file 1:Figs. S5(a) and (b)). With 0.6e, 0.8e and 1e doping, the PMA (perpendicular magnetic anisotropy energy) is 1.70, 2.42, and 5.13 meV, respectively, large enough for spintronic applications (Fig. 8b).
Additionally, Mn3 Br8 monolayer with carrier doping of –1e ~ 1e per formula unit maintains to be half-metallic. Its band gap in the semiconducting spin-channel increases/decreases slightly with the increasing electron/hole doping as shown in Fig. 8d; the positions of the VBM and CBM do not change. Exceptional, Mn3 Br8 monolayer turns to be FM spin-gapless semiconductors (SGS) with the metallic spin-channel opening up a very small energy gap (0.07 eV) under –0.6e and –0.8e hole doping; its Fermi level locates in the band gap region (Fig. 11b and c, more clearly figures are presented in Additional file 1:Figs. S6(a) and (b)). Correspondingly, electrons may be easily excited from the valence band to the conduction band with a small input of energy, which simultaneously produces 100% spin polarized electron and hole carriers. The Curie temperature at –0.6e and –0.8e hole doping is 110 K (Fig. 9c and d), higher than liquid-nitrogen temperature (77 K). Considering with that the charge density modulation of \(10^{13} \sim10^{15} {\text{cm}}^{ - 2}\) was already achieved experimentally [80,81,82], our predicted properties of Mn3 Br8 monolayer with carrier doping are also experimentally approachable.

a - j Spin-resolved band structure for Mn3 Br8 monolayer with carrier doping from -1e to 1e per formula unit. Positive and negative values represent the electron and hole doping, respectively. The green arrow denotes the indirect band gap
Conclusões
In summary, the stability, electronic, and magnetic properties of Mn3 Br8 monolayer have been carefully investigated. Our results show that Mn3 Br8 monolayer is FM half-metal with 130 K Curie temperature and with 2.97 eV band gap for the semiconducting spin-channel. Plus, the magnetic moment of each Mn ion is 13/3μB; the MAE is –2.33 meV per formula unit. The Mn3 Br8 monolayer is designed by inducing single Mn vacancy in the \({2} \times {2} \times {1}\) supercell of MnBr2 monolayer to break the AFM coupling d 5 configuration. The feasibility of forming the Mn vacancy and the dynamical, mechanical stability of Mn3 Br8 monolayer have been comprehensively confirmed. Additionally, Mn3 Br8 monolayer under biaxial strain –5% ~ 5% is still FM half-metal with 2.71 ~ 3.12 eV band gap for the semiconducting spin-channel, whose Curie temperature under –5% biaxial strain is 160 K. Both biaxial strain and carrier doping make the MAE increase, which turns to be perpendicular to the plane under electron doping. With 0.8e and 0.6e hole doping, Mn3 Br8 monolayer turns to be spin-gapless semiconductor (SGS) with band gap of 0.07 eV. Our calculations demonstrate Mn3 Br8 monolayer as FM half-metal with high Curie temperature, and having large MAE and large magnetic moment, and tunable electronic and magnetic properties under the applied biaxial strain and carrier doping.
Disponibilidade de dados e materiais
Os conjuntos de dados gerados durante e / ou analisados durante o estudo atual estão disponíveis junto ao autor correspondente mediante solicitação razoável.
Abreviações
- 2D:
-
Bidimensional
- AFM:
-
Antiferromagnetic
- CBM:
-
Banda de condução mínima
- DFT:
-
Teoria da densidade funcional
- DOS:
-
Densidade de estados
- FIM:
-
Ferrimagnetic
- FM:
-
Ferromagnético
- GGA:
-
Aproximação de gradiente generalizado
- GKA:
-
Goodenough–Kanamori–Anderson
- MAE:
-
Magnetic anisotropy energy
- MCE:
-
Magneto-crystalline anisotropy energy
- MC:
-
Monte Carlo
- MDE:
-
Magnetic dipolar anisotropy energy
- MTJ:
-
Magnetic tunneling junctions
- NM:
-
Não magnético
- NN:
-
Nearest neighboring
- PAW:
-
Projector augmented wave
- PBE:
-
Perdew–Burke–Ernzerhof
- PMA:
-
Perpendicular magnetic anisotropy energy
- PNGN:
-
Porous nitrogen-doped graphene networks
- SGS:
-
Spin-gapless semiconductor
- SOC:
-
Spin–orbit coupling
- VASP:
-
Vienna ab-initio simulation package
- VBM:
-
Banda de valência máxima
- VDW:
-
Van der Waals
Nanopartículas multifuncionais aumentadas por ultrassom com foco de baixa intensidade para a integração de imagens de ultrassom e terapia sinérgica de câncer de mama metastático
Otimização numérica para a configuração geométrica de cerâmicas em compósitos resistentes ao desgaste HCCI / ZTAP com base no modelo de partícula real
Nanomateriais
- Entrevista com Craig Trevor da Persuasion Inc.
- Do piloto à implantação em grande escala:Indo longe com a IoT
- A Bifurcação de Suscetibilidade Magnética no Isolador Topológico Sb2Te3 Ni-Dopado com Ordem Antiferromagnética Acompanhada por Alinhamento Ferromagnético Fraco
- Emissão multicolor da estrutura ultravioleta de nanopiramida quasicristal fotônica baseada em GaN com InxGa1 semipolar − xN / GaN vários poços quânticos
- Absorvedor perfeito de banda larga com monocamada MoS2 e matriz hexagonal de nitreto de titânio nanodisco
- Microesferas de carbono magnético como um adsorvente reutilizável para remoção de sulfonamida da água
- Resposta fotovoltaica pronunciada do fototransistor MoTe2 multicamadas com formulário de contato assimétrico
- Evolução da área de contato com carga normal para superfícies rugosas:de escalas atômicas a macroscópicas
- Otimização da absorção de banda larga e multibanda de grafeno monocamada em frequências ópticas de ressonâncias de dipolo magnético múltiplo em metamateriais
- Remoção de antibióticos da água com uma membrana de nanofiltração totalmente em carbono 3D