


Síntese ionotérmica de silício nanoporoso cristalino e seu uso como materiais de ânodo em baterias de íon-lítio
Resumo
O silício tem um grande potencial como material anódico para baterias de íons de lítio (LIBs) de alto desempenho. Este trabalho relata uma abordagem fácil, de alto rendimento e escalonável para preparar silício nanoporoso, em que siliceto de magnésio comercial (Mg 2 Si) reagiu com o líquido iônico ácido a 100 ° C e à pressão ambiente. O silício obtido consiste em uma estrutura cristalina porosa com uma área superficial BET de 450 m 2 / ge tamanho de poro de 1,27 nm. Quando revestidos com a camada de carbono dopada com nitrogênio e aplicados como ânodo LIB, os compósitos nanoporosos de silício-carbono obtidos exibem uma alta eficiência Coulombic inicial de 72,9% e possuem uma capacidade específica de 1000 mA h g −1 em 1 A g −1 após 100 ciclos. Este método de preparação não envolve vasos de alta temperatura e pressão e pode ser facilmente aplicado para a produção em massa de materiais de silício nanoporoso para bateria de íon-lítio ou para outras aplicações.
Introdução
O consumo cada vez maior e a alta dependência de energia fóssil na sociedade contemporânea têm causado uma crescente sensação de mal-estar sobre o meio ambiente, o clima e o suprimento de energia. Há uma demanda urgente para o desenvolvimento de dispositivos e sistemas sustentáveis, portáteis de alta energia e alta densidade de energia para resolver a fonte de energia temporal e a incompatibilidade ambiental para estilos de vida modernos [1]. As baterias recarregáveis de íons de lítio (LIBs) são uma promessa notável para dispositivos de armazenamento de energia devido à sua densidade de energia relativamente alta e estabilidade de ciclo longo [2, 3]. Para atender aos requisitos crescentes de LIBs de alto desempenho, vários materiais de eletrodo de alta capacidade estão sendo amplamente desenvolvidos, como materiais carbonosos amorfos porosos [4, 5], compósitos à base de fósforo [6, 7], compósitos à base de silício [8 ] e óxidos de metais de transição [9, 10]. Como um componente vital, o silício (Si) é um dos materiais anódicos mais impressionantes devido à sua grande capacidade teórica (4200 mAh g −1 ), fontes naturais abundantes e tensão de captação de lítio relativamente segura [11]. No entanto, a comercialização prática em grande escala de material anódico de silício é atormentada por dois problemas intrincados. Por outro lado, a enorme expansão e contração volumétrica nos processos de carga e descarga levam à quebra do material ativo de silício, rápido desbotamento da capacidade irreversível da bateria [12]. Por outro lado, a baixa eletrocondutividade intrínseca (1,6 × 10 −3 S / m) de silício elementar também impede bastante a transferência de elétrons e diminui a capacidade de velocidade do eletrodo.
Recentemente, esforços consideráveis têm sido concentrados em contornar os problemas de estabilidade mencionados acima [13]. Um grande número de materiais de silício nanoestruturados, incluindo nanotubos [14], nanofios / nanobastões [15, 16] e nanofolhas [17,18,19], foram projetados para alcançar integridade estrutural aprimorada e desempenho de ciclo. Além disso, a preparação de compósitos porosos à base de Si também é considerado um método eficaz, pois os espaços de poros apropriados em compósitos de silício poroso podem atuar como tampões para mitigar a expansão do volume e, assim, melhorar o desempenho de ciclagem em LIBs [20, 21]. Por exemplo, Kim et al. fabricou partículas de silício poroso tridimensional por recozimento térmico e corrosão géis de Si revestidos com butil e SiO 2 nanopartículas a 900 ° C sob uma atmosfera de Ar, que exibiu uma capacidade estável de mais de 2800 mA h g −1 após 100 ciclos a 1 ° C [22]. An et al. relataram uma via verde, escalável e controlável para preparar silício nanoporoso (NP-Si) com excelentes propriedades eletroquímicas de Mg 2 comercial Liga de Si via destilação a vácuo de alta temperatura [23]. Embora avanços tremendos no desempenho eletroquímico consumado tenham sido demonstrados, a maioria dos métodos de preparação para essas estruturas nanoporosas de Si são geralmente muito complicados para aumentar a escala.
Outra tática eficaz para impulsionar o desempenho eletroquímico do ânodo de silício é revestir carbono eletronicamente condutor em partículas de nanosilício para formar nanocompósitos de silício-carbono [19, 24], como casca de gema [25], melancia [26] e estruturas ocas [ 27]. Por exemplo, Pan et al. projetou nanocompósitos de Si – C estruturados em casca de gema com alta capacidade específica e boa estabilidade de ciclo por um método simples e de baixo custo baseado na tecnologia de corrosão de NaOH [28]. Chen et al. desenvolveu um Si / B estruturado com núcleo-shell 4 Compósito C com revestimento de grafite e demonstrou que tais compósitos possuíam boa estabilidade de ciclo de longo prazo [29]. Vários estudos demonstraram que o carbono condutivo pode não apenas compensar a baixa condutividade elétrica do silício, mas também servir como um intermediário elástico para retardar a grande mudança de volume e prevenir o contato direto entre os materiais ativos de silício e o eletrólito, levando a uma maior estabilidade do ciclo [30].
Até o momento, as rotas sintéticas para nanopartículas de silício (Si NPs) ou silício poroso (pSi) geralmente envolvem decomposição térmica de silanos [31], corrosão química de pastilhas de Si e redução magnesiotérmica de SiO 2 modelos [32, 33]. Essas preparações geralmente requerem várias etapas, alta temperatura, modelos de custo relativamente alto, etc., o que leva a alto custo e dificuldades para aumentar a escala [34]. Recentemente, a preparação de Si NPs em solução também tem recebido muita atenção [35, 36]. Por exemplo, Kauzlarich et al. relatou que SiCl 4 reagiu com NaSi ou KSi em solventes orgânicos para obter nanopartículas de silício [37]. Liang et al. preparou as nanoesferas de silício semelhantes a ninhos por meio de uma reação solvotérmica, na qual NaSi reagiu com NH 4 Br no solvente misto de piridina e dimetoxietano em autoclave a 80 ° C por 24 h [38]. A síntese de solução relatada geralmente envolveu agentes redutores altamente ativos, como metais alcalinos, LiAlH 4 e NaSi e freqüentemente produziam baixos rendimentos ou pequenas quantidades de Si NPs. Nesse sentido, para a fabricação em massa de nanossilício, uma abordagem simples, escalonável e de baixo custo ainda é imperativa. Aqui, apresentamos uma preparação conveniente de alto rendimento de silício poroso por oxidação de Mg 2 Si em líquido iônico ácido a 100 ° C e pressão ambiente. Quando revestido com uma camada de carbono dopado com nitrogênio e servido como ânodo de bateria de íon-lítio, os compósitos nanoporosos de silício-carbono obtidos exibiram uma alta eficiência coulômbica inicial (CE) de 72,9% e entregaram uma capacidade específica de 1000 mA h g -1 após 100 ciclos a 1 A g −1 .
Métodos
Materiais
Cloreto de 1-butil-3-metilimidazólio ([Bmim] Cl) foi fornecido por Shanghai Cheng Jie Chemical Co. LTD. Cloreto de alumínio (AlCl 3 ) foi adquirido na Sinopharm Chemical Reagent Co., Ltd. Siliceto de magnésio (Mg 2 Si) e pó de silício comercial (1–5 µm) foram comprados da Alfa Aesar. Carbonato de etileno (EC), carbonato de dietila (DEC), carbonato de fluoroetileno (FEC) e LiPF 6 foram adquiridos de Shenzhen Kejingstar Technology Ltd., China. Todos os produtos químicos e reagentes foram usados diretamente conforme recebidos.
Síntese de nanopartículas de silício poroso (pSi)
Em um procedimento típico, [Bmim] Cl (1,5 g) e AlCl 3 (4,5 g) com uma razão molar de ~ 1:4 foram misturados e carregados em um tubo de vidro Schlenk. Posteriormente, 500 mg de siliceto de magnésio (Mg 2 Si) foram adicionados ao tubo de vidro e agitados vigorosamente a 100 ° C durante 10 h. O procedimento acima foi conduzido em um porta-luvas cheio de Ar. Após arrefecimento, o precipitado foi recolhido e lavado com ácido clorídrico 1 M, água destilada e etanol. Finalmente, o produto (150 mg, rendimento de 82%) foi seco em vácuo para posterior caracterização.
Síntese de carbono dopado com nitrogênio revestido em nanopartículas de silício poroso (pSi @ NC)
O procedimento de preparação é referido às literaturas relatadas [39, 40]. Primeiro, 0,1 g das nanopartículas de silício poroso obtidas (pSi) foram dispersas em 250 mL de água desionizada contendo dodecilbenzenossulfonato de sódio (SDBS; 5 mg) por ultrassonicação por 30 min. A mistura foi agitada vigorosamente durante 1 h à temperatura ambiente. Depois disso, 200 μL de monômero de pirrol, 0,34 g de (NH 4 ) 2 S 2 O 8 , e 1,25 mL de HCl 1 M foram adicionados à solução acima. Após a mistura ter sido agitada em um banho de gelo / água por 24 h, os pós pretos formados (denotados como pSi @ PPy) foram reunidos por filtração, lavados com água desionizada e secos em vácuo. Finalmente, a amostra pSi @ PPy foi aquecida a uma taxa de rampa de 5 ° C min −1 em um forno tubular a 700 ° C por 3 h em uma atmosfera fluida de Ar para obter o compósito pSi @ NC. O teor de carbono foi estimado por estudos termogravimétricos.
Medições eletroquímicas
As propriedades eletroquímicas das nanopartículas de silício poroso foram estudadas usando uma meia célula tipo moeda CR2032, na qual folhas metálicas de lítio serviram como contra-eletrodos e eletrodo de referência, o pSi @ NC como preparado como eletrodo de trabalho, filmes macroporosos de polipropileno (Celgard 2400) como separadores e 1,0 M LiPF 6 em mistura 1:1 (v / v) de carbonato de etileno (EC) / carbonato de dietila (DEC) como eletrólito. As células CR2032 foram montadas em uma caixa de luvas com atmosfera de argônio (conteúdo de oxigênio e água inferior a 0,1 ppm). Os eletrodos do ânodo de trabalho foram preparados pela mistura do compósito pSi @ NC obtido, carbono super P e alginato de sódio em uma proporção em peso de 70:20:10 em água desionizada para formar uma pasta homogênea. Em seguida, a pasta foi revestida em folha de Cu e seca sob condição de vácuo a 80 ° C por 12 h. A massa total de carga dos materiais ativos no eletrodo foi de aproximadamente 0,5 mg cm −2 . Os ciclos de carga e descarga das meias-células foram realizados em um testador de bateria Neware (Shenzhen, China) em um modo de corrente constante na faixa de 0,01-1,5 V. A voltametria cíclica (CV) dos ânodos preparados foi medida em uma estação de trabalho eletroquímica CHI650d (Shanghai Chenhua Instruments Inc., China), usando uma célula de três eletrodos com a taxa de varredura de tensão de 0,2 mV s −1 à temperatura ambiente. A capacidade específica foi calculada com base na massa total dos compósitos pSi @ NC.
Métodos de caracterização
As medições de difração de raios-X de potência (PXRD) foram realizadas em um difratômetro de raios-X Bruker D8 ADVANCE (radiação Cu Kα, 40 kV, 40 mA, λ =1,5418 Å). A morfologia e a microestrutura das amostras foram obtidas por microscopia eletrônica de varredura (microscópio eletrônico de varredura por emissão de campo Hitachi, S-4800), e a espectroscopia de dispersão de energia foi utilizada para analisar a distribuição elementar. Microscopia eletrônica de transmissão (TEM) e imagens TEM de alta resolução foram registradas em um equipamento JEM-2100. Os parâmetros porosos foram determinados usando um analisador Micromeritics ASAP 2020 a 77 K após a desgaseificação da amostra a 150 ° C por 10 h. A área de superfície específica foi calculada usando o método Brunauer − Emmett − Teller (BET) de múltiplos pontos, e a distribuição do tamanho dos poros foi analisada pelo método da teoria funcional da densidade (DFT) com base nos dados de adsorção. A espectroscopia Raman (LabRAM Aramis, Horiba, equipada com um laser de comprimento de onda de 633 nm) foi usada para investigar a estrutura do silício nanoporoso, que foi primeiro calibrado com um wafer de Si (520 cm −1 ) O espectrômetro PHI 5000 VersaProbe foi usado para medições de espectroscopia de fotoelétrons de raios-X (XPS). A análise termogravimétrica (TGA) foi conduzida em um analisador térmico STA449F3 (Netzche) simultâneo sob atmosfera de ar a 10 ° C min −1 de 30 a 800 ° C no fluxo de ar. Os testes de voltametria cíclica (CV) foram realizados em uma estação eletroquímica CHI650d (Shanghai Chenhua Instruments Inc., China).
Resultados e discussão
A preparação de nanopartículas de silício poroso (pSi) a partir de Mg 2 Si em líquido iônico pode ser expresso como Reação 1, como mostrado no Esquema 1. Para entender o processo de reação, os produtos puros da Reação 1 proposta sem qualquer tratamento de lavagem foram diretamente coletados e analisados por PXRD (Arquivo adicional 1:Figura S1) . A análise PXRD mostrou que o produto puro era composto principalmente de Si cristalino, subproduto de sais inorgânicos MgCl 2 e reagentes Mg 2 Si e AlCl 3 . No processo de preparação de nanopartículas de silício porosas, cloreto de 1-butil-3-metilimidazol e tricloreto de alumínio em uma razão molar de 1:4 foram misturados para garantir que o sistema de reação seja ácido. Então, Mg 2 O Si reagiu com o sistema ácido para formar as nanopartículas de silício. O rendimento de nanopartículas de silício poroso foi superior a 82% com base na quantidade de átomos de Si em Mg 2 Si. A reação foi realizada em um frasco, proporcionando uma produção em massa de pSi facilmente escalonável. O uso de líquido iônico [BmimCl] -AlCl 3 foi necessário para a preparação do pSi. Sem AlCl 3 , a reação de Mg 2 Si com [BmimCl] não pôde ocorrer. Da mesma forma, o Mg 2 Si não pôde reagir com AlCl 3 sozinho ou em outros solventes orgânicos, como tetra-hidrofurano, para produzir pSi. Notamos que o pSi foi previamente preparado por decomposição térmica de silanos ou halogenetos de silício em alta temperatura, ou suas reações com agentes redutores altamente ativos, como metais alcalinos, LiAlH 4 e NaSi [37, 41]. O uso de Mg 2 O Si na preparação de silício nanoestruturado por destilação do Mg em alta temperatura também é conhecido [23, 42, 43]. No entanto, essas reações frequentemente produziram baixos rendimentos ou pequenas quantidades de pSi. Em contraste, o método relatado neste trabalho é aplicável para a produção em massa de pSi.
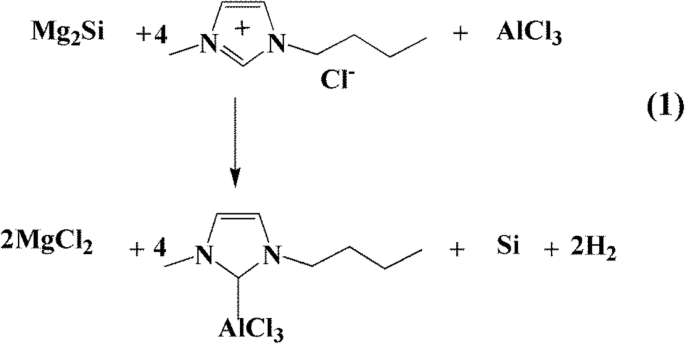
A reação proposta para a preparação de pSi
O padrão PXRD do produto é mostrado na Fig. 1a. Estes cinco picos estreitos e agudos em 2θ 28,4, 47,3, 56,1, 69,1 e 76,4 ° são atribuídos aos planos de rede (111), (220), (311), (400) e (331) da fase de silício cúbico (JCPDS No. 27-1402), o que sugere que o silício obtido é altamente cristalino. Os tamanhos médios de cristalito das partículas de silício obtidas eram de cerca de 40 nm com base na equação de Scherrer. A Figura 1b mostra os espectros Raman das nanopartículas de silício. O pico característico típico localizado em torno de 518 cm −1 corresponde ao modo de alongamento Si-Si do Si cristalino. A banda larga entre 900 e 1050 cm −1 deve ser atribuído ao espectro de segunda ordem do silício [44]. E o pequeno pico em ~ 303 cm −1 foi atribuído ao óxido de superfície. As áreas de superfície específicas e caracterização da porosidade das amostras obtidas foram elucidadas por N 2 isotermas de adsorção / dessorção a 77 K. A amostra de pSi exibiu curvas de sorção isotérmica tipo IV (a) com uma alça de histerese híbrida H2 (b) / H3, que é característica de um material de estrutura porosa [45]. Ele possuía uma alta área de superfície Brunauer-Emmett-Teller (BET) de 450 m 2 g −1 . A análise de distribuição de tamanho de poro com base no método DFT mostrou que o produto consistia em microporos relativamente estreitos (1,27 nm) e mesoporos (5,4 nm) com ampla distribuição de tamanho de poro. A presença desses poros pode facilitar Li + difusão de íons.
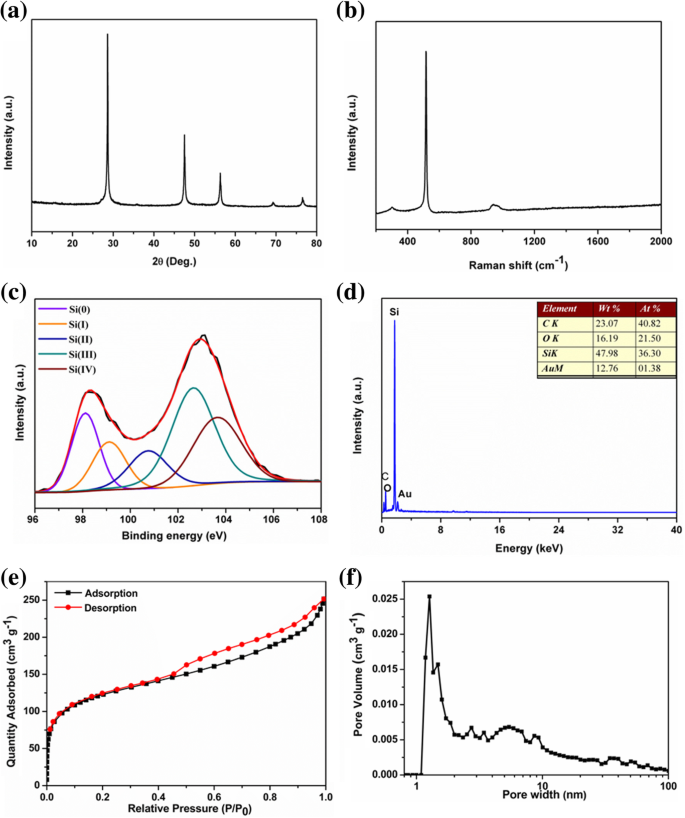
a Padrões PXRD, b Espectro Raman, c Espectro XPS, d Espectro EDS, e curvas de adsorção-dessorção de nitrogênio e f a curva de distribuição de tamanho de poro de pSi
A morfologia das amostras de silício obtidas foi estudada por meio de microscopia eletrônica de varredura (MEV) e microscopia eletrônica de transmissão (TEM). As imagens SEM (Fig. 2a, b, Arquivo adicional 1:Figura S2) e TEM (Fig. 2c, d) mostram que os tamanhos de partícula gerais das partículas nanoporosas de silício obtidas variam de várias dezenas a cerca de 100 nm de diâmetro. A imagem TEM na Fig. 2c mostra que a amostra é composta de partículas de silício interconectadas, resultando em uma estrutura porosa. Postulamos que o arranjo estreito de Si 4− no precursor de tamanho mícron 2 O Si reagiu com o líquido iônico ácido para formar Si rodeado por MgCl 2 nanopartículas. Os últimos foram lavados por HCl diluído, deixando pSi interconectado com as vagas. O pSi obtido mostrou uma grande área de superfície BET de 450 m 2 g −1 com distribuição uniforme de tamanho de poro em 1,27 nm, apoiando a postulação acima. A imagem HRTEM de pSi na Fig. 2d revela que a franja da rede clara com um típico d O espaçamento de 0,31 nm, atribuído aos (111) planos cristalinos do Si cúbico, está de acordo com os resultados do PXRD. As nanopartículas de silício interconectadas mostram-se recobertas por uma fina camada de óxido na superfície externa, devido à oxidação. A composição da superfície e o estado de valência das nanopartículas de Si foram identificados por análise de dispersão de energia (EDS) e espectroscopia de fotoelétrons de raios-X (XPS). O espectro de Si 2p XPS (Fig. 1c) mostrou dois picos amplos e sobrepostos a 98,2 eV e 103,0 eV. Os dois picos podem ser divididos em cinco componentes em 98,11, 99,11, 100,75, 102,64 e 103,64 eV, que foram atribuídos a Si (0), Si (I), Si (II), Si (III) e Si (IV ), respectivamente. A presença de um pico forte de Si (0) implica na formação de silício poroso. Os picos mais fortes de Si (III) e Si (IV) sugerem que a superfície do silício poroso foi revestida por óxido de silício [46]. Consistentemente, a análise de energia dispersiva (EDS) de pSi mostrou que a razão atômica de Si / O na superfície era de cerca de 3:2 (Fig. 1d).

a , b Imagens SEM e c , d Imagens TEM de pSi (a inserção em d mostra os padrões SAED)
Para serem usados como materiais anódicos LIB, os pSi foram encapsulados com polipirrol condutor para formar compósitos pSi @ NC. O padrão PXRD do compósito pSi @ NC mostrou um pico largo adicional em torno de 23 ° (Fig. 3a), sugerindo que a camada de carbono dopada com nitrogênio é amorfa [39]. O espectro Raman do composto pSi @ NC (Fig. 3b) mostrou dois picos largos em 1335 e 1585 cm -1 atribuído às bandas D e G do carbono grafítico [47], respectivamente, o que confirma o resultado PXRD. A relação de intensidade da banda D e banda G (I D / I G ) do compósito pSi @ NC é de cerca de 1,07, implicando em um baixo grau de grafitização da camada de carbono. Os espectros C 1 s XPS de pSi @ NC mostraram a existência da ligação N – C (285,85 eV na Fig. 3c), confirmando que o nitrogênio foi dopado na estrutura de carbono [48]. O pico N 1 s XPS (Fig. 3d) pode ser dividido em três picos centralizando em 397,85, 398,72 e 400,57 eV, respectivamente, que pertencem aos tipos piridínico, pirrólico e grafítico de átomos de nitrogênio dopados na estrutura de carbono [39 , 49]. O teor de carbono no compósito pSi @ NC foi determinado por TGA como sendo cerca de 20% em peso (Arquivo adicional 1:Figura S3).
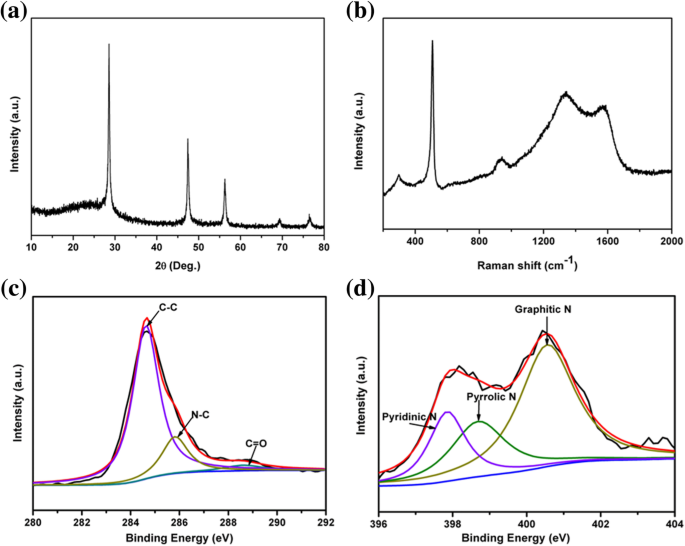
a Padrões PXRD, b Espectro Raman, c espectros C 1 s XPS de alta resolução e d espectros N 1 s XPS de alta resolução do composto pSi @ NC
Para caracterizar os desempenhos eletroquímicos do compósito pSi @ NC como o ânodo de LIBs, medições de voltametria cíclica (CV) entre e 2,5 V a uma taxa de varredura de 0,2 mV s −1 foram realizados. Conforme mostrado na Fig. 4a, o primeiro pico de redução em torno de 1,5 V nas curvas CV foi atribuído à decomposição do aditivo eletrolítico (carbonato de fluoroetileno FEC) [50]. O pico de redução irreversível era visível no potencial em torno de 0,6 V durante a primeira descarga e desaparecia nos ciclos subsequentes, o que estava associado à geração da membrana de interface de eletrólito sólido (SEI) [51]. A formação de SEI foi devido à decomposição de solventes orgânicos de eletrólitos como EC e DEC e levou à perda de capacidade irreversível inicial [50, 52]. O pico próximo a 0,1 V nas próximas curvas CV representou a transição do silício cristalino para Li amorfo x Si [53]. Enquanto isso, durante o processo de carga, dois picos redox típicos em torno de 0,28 e 0,53 V foram observados, os quais estavam relacionados ao processo de extração de Li de Li x Si [54, 55]. Notavelmente, as intensidades atuais dos picos anódicos e catódicos aumentaram gradualmente após os primeiros ciclos. Este fenômeno de “ativação” deve ser atribuído principalmente à degradação gradual da estrutura cristalina do silício [54, 56].
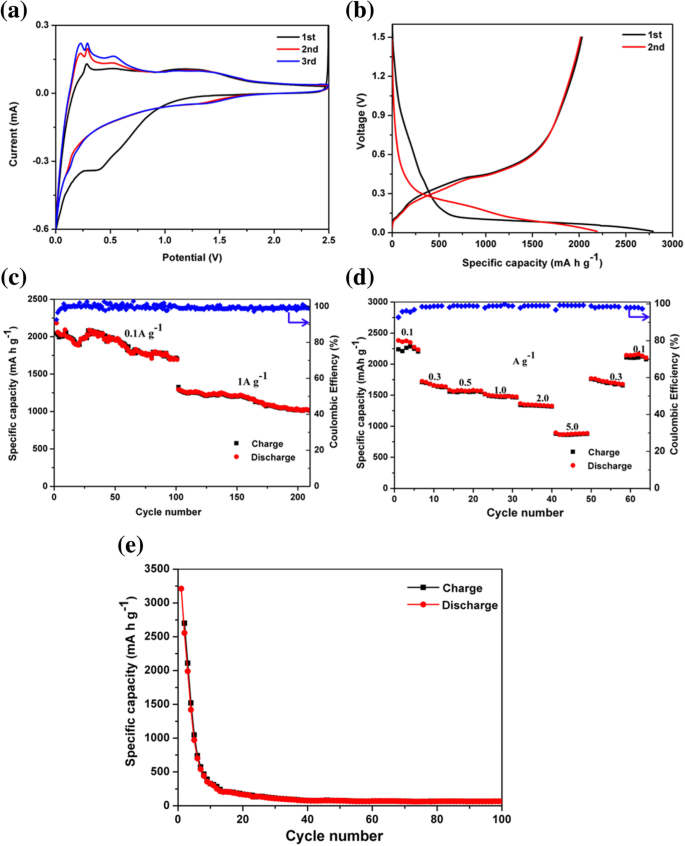
a Curvas CV, b curvas de carga-descarga, c desempenho de ciclismo de longo prazo a 0,1 A g −1 e 1 A g −1 por 100 ciclos, respectivamente (densidades de corrente), e d taxa de desempenho ciclada em várias densidades de corrente do eletrodo composto pSi @ NC. e Desempenho de ciclagem do composto comercial Si @ NC a 0,1 A g −1 por 100 ciclos
A Figura 4b ilustra as duas primeiras curvas descarga-carga dos ânodos compostos pSi @ NC circulando a uma densidade de corrente de 0,1 A g −1 . O compósito pSi @ NC teve um terraço de descarga longo e plano em torno de 0,1 V durante a primeira descarga, o que está de acordo com o terraço característico das inserções de Li de Si cristalino. O silício bem cristalizado tornou-se amorfo e mostrou os perfis representativos de carga / descarga do silício amorfo nos ciclos subsequentes. Os outros planaltos potenciais que surgiram em torno de 0,6 V durante o primeiro processo de litiação resultaram da formação do SEI [57]. Os resultados estão de acordo com as curvas CV. A descarga inicial e as capacidades de carga foram 2790 e 2036 mA h g −1 , entregando uma alta eficiência Coulombic inicial (CE) de 72,9%. A capacidade de carga mais baixa pode ser parcialmente devido ao efeito de restrição da camada de óxido SiO x , que serviu como amortecedores para limitar a expansão do volume e a extensão da litiação [58, 59]. É importante ressaltar que nenhuma queda de capacidade óbvia foi observada nos ciclos subsequentes, e a eficiência Coulombic foi mantida quase constante em torno de 100%.
A Figura 4c mostra o desempenho cíclico dos ânodos de compósitos pSi @ NC, que foram conduzidos a uma densidade de corrente de 0,1 A g −1 por 100 ciclos e com uma densidade de corrente de 1 A g −1 para os 100 ciclos subsequentes. Os ânodos nanocompósitos pSi @ NC mostraram uma capacidade de 1720 mA h g −1 após 110 ciclos a uma densidade de corrente de 0,1 A g −1 , correspondendo a uma retenção de capacidade de 79%. Além disso, os eletrodos de compósito pSi @ NC forneceram uma capacidade reversível de 1010 mA h g −1 em 1 A g −1 após 110 ciclos subsequentes, com uma taxa de decaimento de capacidade de 0,2% por ciclo de 101 a 210º ciclo. A Figura 4d mostra o desempenho da taxa do eletrodo pSi @ NC. O eletrodo pSi @ NC alcançou capacidades de descarga de 2360, 1690, 1570, 1470, 1320 e 850 mA h g −1 na densidade de corrente de 0,1, 0,3, 0,5, 1,0, 2,0 e 5,0 A g −1 , respectivamente. A capacidade de descarga pode ser recuperada para aproximadamente 2160 mA h g −1 quando a densidade da corrente voltou a 0,1 A g −1 , provando que o ânodo composto pSi @ NC tinha uma reversibilidade eletroquímica notável. Em comparação, o pó de silício comercial (Fig. 4e) revestido com o carbono condutor dopado com nitrogênio como um ânodo atingiu uma alta capacidade de descarga inicial de 3230 mA h g −1 , mas sofreu severa deterioração da capacidade para 110 mA h g - 1 após 100 ciclos a 0,1 A g −1 . Estes resultados sugeriram que a camada de carbono dopada com nitrogênio e a estrutura porosa em pSi @ NC poderiam fornecer as vias de transporte rápido de íons / elétrons e manter a estabilidade estrutural, dotando assim o ânodo de compósito pSi @ NC de boa taxa de desempenho e excelente reversibilidade [ 21, 39, 60]. Além disso, a oxidação da superfície em pSi também pode contribuir para melhorar a eficiência do ciclo das baterias de íon-lítio, o que limitou a expansão do volume das partículas de silício e evitou algumas reações colaterais de acordo com os estudos anteriores [58].
Conclusões
Em resumo, desenvolvemos um novo método para preparar silício nanoporoso em alto rendimento com base na reação de siliceto de magnésio (Mg 2 Si) em líquido iônico ácido. Quando revestidos com a camada de carbono dopada com nitrogênio e aplicados como um ânodo de bateria de íon-lítio, os compostos de silício-carbono obtidos exibiram alta capacidade reversível, estabilidade de ciclo de longo prazo e alta eficiência columbica inicial. A camada de revestimento de carbono dopada com N forneceu caminhos condutores eficientes para o transporte rápido de íons de lítio e transferência de elétrons, o que é benéfico para aumentar as propriedades eletroquímicas das partículas de silício. Uma vez que a condição da reação é relativamente amena e o rendimento dos produtos é superior a 82%, este método de preparação pode ser estendido à produção em massa de materiais de anodo de silício.
Abreviações
- [Bmim] Cl:
-
Cloreto de 1-butil-3-metilimidazólio
- AlCl 3 :
-
Cloreto de alumínio
- CV:
-
Voltametria cíclica
- EDS:
-
Espectroscopia de dispersão de energia
- Mg 2 Si:
-
Siliceto de magnésio
- pSi:
-
Nanopartículas de silício poroso
- pSi @ NC:
-
Carbono dopado com nitrogênio revestido em nanopartículas de silício poroso
- PXRD:
-
Difração de raios-x de pó
- SEM:
-
Microscopia eletrônica de varredura
- TEM:
-
Microscopia eletrônica de transmissão
- TGA:
-
Análise termogravimétrica
- XPS:
-
espectroscopia de fotoelétrons de raios-X
Tinta Nano-prata de alta condutividade e baixa temperatura de sinterização para eletrônicos de papel
Usando nanopartículas magnéticas carregadas positivamente para capturar bactérias em concentração ultralow
Nanomateriais
- Composto Híbrido de Sílica Nanoestruturada / Ouro-Celulose Ligado Amino-POSS via Processo Sol-Gel e Suas Propriedades
- Síntese fácil de nanopartículas de SiO2 @ C ancoradas em MWNT como materiais de ânodo de alto desempenho para baterias de íon-lítio
- Síntese e propriedades eletroquímicas de materiais catódicos de LiNi0,5Mn1,5O4 com dopagem composta Cr3 + e F− para baterias de íon-lítio
- Composto de MoS2 / Acetileno com poucas camadas como um ânodo eficiente para baterias de íon-lítio
- Preparação de micrromateriais híbridos MnO2 revestidos com PPy e seu desempenho cíclico aprimorado como ânodo para baterias de íon-lítio
- Composto de Si / Grafeno incorporado fabricado por redução térmica de magnésio como material de ânodo para baterias de íon-lítio
- Síntese fácil e ecológica de nanofios de Co3O4 e sua aplicação promissora com grafeno em baterias de íon-lítio
- Síntese sonoquímica fácil de uma etapa e propriedades fotocatalíticas de compostos de pontos quânticos de grafeno / Ag3PO4
- Um ânodo de filme Fe2O3 nanocristalino preparado por deposição de laser pulsado para baterias de íon-lítio
- Síntese rápida de nanocristais de Pt e materiais La2O3 Pt / microporosos usando levitação acústica