


Strain Tunable Bandgap e High Carrier Mobility em SiAs e SiAs2 Monocamadas de Estudos de Primeiros Princípios
Resumo
A pesquisa de novos materiais bidimensionais (2D) atomicamente finos e estáveis e autônomos é de grande interesse nos aspectos fundamentais e práticos das ciências de materiais contemporâneas. Recentemente, foi realizada a síntese de monocristais de SiAs em camadas, o que indica que sua estrutura de poucas camadas pode ser esfoliada mecanicamente. Executando cálculos de teoria funcional de densidade de primeiros princípios, propusemos dois SiAs e SiAs semicondutores estáveis dinamicamente e termodinamicamente 2 monocamadas. O cálculo da estrutura de banda revela que ambos exibem lacunas de banda indiretas e uma transição de banda indireta para direta até mesmo para metal é encontrada pela aplicação de deformação. Além disso, descobrimos que SiAs e SiAs 2 monocamadas possuem mobilidade de portadora muito maior do que MoS 2 e exibir transporte anisotrópico como o fosforeno preto, tornando-os aplicação potencial em optoeletrônica. Nossos trabalhos abrem uma nova rota em nanoescala para novas funcionalidades de dispositivos ópticos.
Histórico
Cristais bidimensionais (2D) atomicamente finos se tornaram um dos campos de mais rápido crescimento da ciência de materiais contemporânea. As propriedades eletrônicas versáteis, a excelente mobilidade de elétrons e as aplicações promissoras em nanoeletrônica e optoeletrônica estão levando uma grande porcentagem de físicos da matéria condensada a buscar novos materiais 2D. Seguindo o grafeno [1-4], um grande número de outros materiais 2D foram sintetizados, como siliceno [5-7], nanofolhas de nitreto de boro [8, 9], dichalcogenetos de metais de transição (TMDs) [10, 11], fósforo preto [12, 13], borofeno [14-16], arseneno [17, 18], telureno [19] e seus compostos isoeletrônicos [20-23]. A lista de materiais 2D está se expandindo rapidamente e mais de milhares de tipos de tais materiais são agora conhecidos, abrangendo todo o espectro de propriedades eletrônicas e outras. E suas novas propriedades, diferentes ou até melhores do que as de suas contrapartes em massa, são teoricamente previstas e experimentalmente confirmadas com firmeza.
Embora esforços extensos e substanciais tenham sido investidos na localização de diversos materiais 2D, incluindo alguns que já possuem bandgaps ou outras propriedades desejáveis, o consenso não foi alcançado. O grafeno com maravilhosa mobilidade de portadores, alta estabilidade mecânica e elétrons dirac sem massa tem atraído muita atenção até agora, mas a falta de um gap intrínseco impede sua aplicação na indústria de dispositivos eletrônicos modernos. Embora grandes esforços tenham sido feitos, a abertura de um gap considerável sem efeito colateral não foi alcançada [24, 25]. TMDs com alto desempenho em dispositivos optoeletrônicos, de fato, têm banda gap intrínseca, mas exibem baixa mobilidade da portadora [26-28]. Fósforo preto e azul com uma lacuna de banda ajustável sensível à deformação e alta mobilidade anisotrópica de portadores não podem se manter estáveis no ar [13, 29]. Recentemente, a síntese de SiAs e SiAs em camadas 2 cristais únicos foram realizados [30–32], o que indica que poucas estruturas de camadas podem ser obtidas por esfoliação mecânica.
No presente trabalho, com base nos cálculos da teoria funcional da densidade de primeiros princípios (DFT), propusemos duas monocamadas semicondutoras dinamicamente e termodinamicamente estáveis SiAs e SiAs 2 . Ambos possuem band gaps indiretos (2,39 eV e 2,13 eV respectivamente). A aplicação de deformação isotrópica ao longo de duas direções no plano transforma praticamente os SiAs (SiAs 2 ) monocamada em um material com folga direta de 1,75 eV (1,60 eV). Além disso, descobrimos que SiAs e SiAs 2 monocamadas possuem mobilidade de portadora muito maior do que MoS 2 e exibir transporte anisotrópico como o fosforeno preto, tornando-os aplicação potencial em optoeletrônica. Nossos trabalhos abrem um novo caminho em nanoescala para novas funcionalidades de dispositivos ópticos.
Métodos Computacionais
Os cálculos DFT são realizados usando o código do pacote de simulação ab initio de Viena (VASP) [33]. Usamos o funcional de correlação de troca de Perdew-Burke-Ernzerhof (PBE) [34] sob a aproximação de gradiente generalizado (GGA). O método de onda aumentada do projetor (PAW) [35] foi empregado para descrever a interação elétron-íon. Um vácuo de 20 Å perpendicular às folhas (ao longo do eixo c) foi aplicado para evitar a interação entre as camadas. Um corte de energia cinética de 500 eV é usado para o conjunto de base da onda plana. A amostragem da zona de Brillouin é realizada com uma grade de Monkhorst-Pack [36] 15 × 5 × 1 para folhas 2D. Os critérios de convergência empregados tanto para o relaxamento eletrônico autoconsistente quanto para o relaxamento iônico são definidos como 10 −4 e 0,01 eV / Å para energia e força, respectivamente. Os cálculos do fônon são realizados usando o método da supercélula através do código PHONOPY [37, 38], e as constantes de força do espaço real das supercélulas são calculadas na teoria de perturbação funcional da densidade (DFPT) implementada no VASP. Além disso, uma energia mais estrita (10 −8 eV / átomo) e critério de convergência de força (10 −4 eV / Å) são usados durante os cálculos dos espectros vibracionais. Nos cálculos de dinâmica molecular (MD), (3 × 3 × 1) supercélulas são empregadas e a temperatura é mantida a 300 K por 6 ps com um intervalo de tempo de 2 fs no conjunto moles-volume-temperatura (NVT). Os espectros raman foram calculados no nível da teoria PBE usando o código CASTEP [39-41].
Resultados e discussões
As estruturas geométricas e a distribuição da densidade eletrônica de SiAs e SiAs 2D independentes e relaxados 2 são apresentados nas Fig. 1a, b, respectivamente, e suas estruturas em massa são mostradas no Arquivo Adicional 1:Figura S1 do material suplementar. Conforme mostrado no arquivo adicional 1:Figura S1a eb, os SiAs em massa (SiAs 2 ) possui simetria C2 / m (Pbam) e consiste em camadas de Si-As empilhadas fracamente ligadas por forças de van der Waals com uma distância de 3,06 Å (1,66 Å). A célula unitária da monocamada SiAs é rômbica e seus parâmetros de cristal otimizados são a 1 =3,69Å e b 1 =10,83Å com φ =99,81 °. SiAs contém 6 átomos de Si e 6 átomos de As. Cada átomo de Si tem quatro átomos vizinhos mais próximos (3 As e 1 Si), enquanto cada átomo de As forma apenas três ligações covalentes com os átomos de Si vizinhos. Existem dois tipos de ligações, a saber, ligações Si – Si e Si – As. E o comprimento da ligação Si – Si é de cerca de 2,35 Å e o de Si – As está na faixa de 2,39 Å e 2,43 Å, e a altura de encurvamento é d 1 =4,86 Å. Na vista lateral de SiAs monocamada, uma estrutura semelhante a um cordão de óculos é formada com camadas duplas e simples alternadamente volumosas. Outra estrutura de monocamada de composto de silício e arsênio é SiAs 2 . Sua célula principal contém 4 átomos de Si e 8 átomos de As, com estrutura retangular e os parâmetros de cristal otimizados são a 2 =3,68Å e b 2 =10,57 Å. Cada átomo de As tem três átomos de Si vizinhos mais próximos ou forma uma ligação covalente com átomos de Si vizinhos e duas ligações covalentes entre si, enquanto cada átomo de Si tem apenas quatro átomos de As vizinhos mais próximos. Ao contrário do anterior, SiAs 2 possui uma ligação As – As mais fraca (2,50 Å) em vez da ligação Si – Si. E suas ligações Si-As variam de 2,41 Å a 2,45 Å, e a altura de dobra é d 2 =5,09 Å. A partir da distribuição da densidade de elétrons, os átomos de As atraem elétrons de átomos de Si por sua grande eletronegatividade e têm uma densidade de elétrons maior. A fim de auxiliar a futura caracterização experimental, nós ainda calculamos e verificamos os espectros Raman de SiAs e SiAs em massa e monocamada 2 . Mudanças claras entre a monocamada e os cristais completos foram vistas no arquivo adicional 1:Figura S2 do material suplementar, cujas origens foram identificadas como a influência da interação de van der Waals das camadas [42].
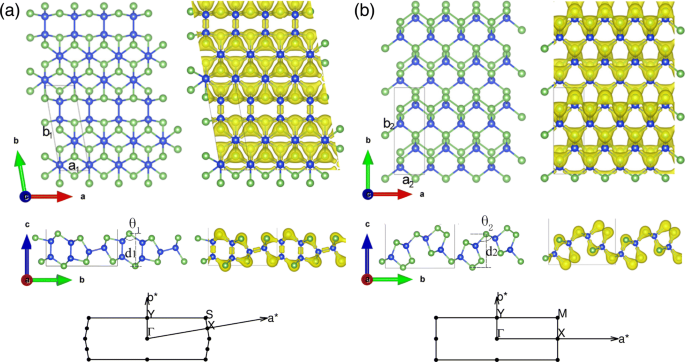
Estrutura geométrica e distribuição de densidade eletrônica de monocamadas SiAs e SiAs 2 . (Cor online) Vistas superior e lateral das monocamadas a SiAs e b SiAs 2 estrutura geométrica e distribuição de densidade de elétrons e a zona de Brillouin associada. As bolas azul e verde indicam os átomos de Si e As, respectivamente
Para aprender a estabilidade dos SiAs (SiAs 2 ), primeiro calculamos a energia coesiva, definida como E coh =( nE Si + mE Como - E Mono ) / ( n + m ), onde E Si , E Como , e E Mono são as energias totais de um único átomo de Si, um único átomo de As e uma unidade de fórmula de monocamada SiAs (SiAs 2 ), respectivamente, e n (m) é o número do átomo de As (Si) na unidade da fórmula. Nossos cálculos mostram que a monocamada de SiAs tem uma energia coesiva de 5,13 eV / átomo, que é um pouco maior do que a de SiAs 2 monocamada 4,98 eV / átomo. Para efeito de comparação, no mesmo nível teórico, as energias coesivas do arseneno e do siliceno são 2,99 e 3,71 eV / átomo, respectivamente [18, 43]. As altas energias coesivas de SiAs e SiAs 2 revelam que ambos estão fortemente ligados com alta estabilidade.
Para confirmar ainda mais as estabilidades estruturais de SiAs e SiAs monocamada 2 , também realizamos cálculos de espectros vibracionais de fônons. Conforme mostrado na Fig. 2a, as frequências positivas são responsáveis pela maioria dos modos, exceto o modo acústico transversal próximo ao Γ ponto, que é devido ao amolecimento dos fônons e foi relatado em outros sistemas semelhantes [44, 45], indicando que as estruturas são dinamicamente estáveis. Em seguida, realizamos simulações de primeiros princípios de 6 ps em temperatura ambiente ( T =300 K ), conforme apresentado na Fig. 2b. A ligeira flutuação de energia e as resistências bem conservadas sugerem que são termicamente estáveis à temperatura ambiente. Nossos resultados implicam que as monocamadas SiAs e SiAs 2 pode ser realizado experimentalmente à temperatura ambiente.
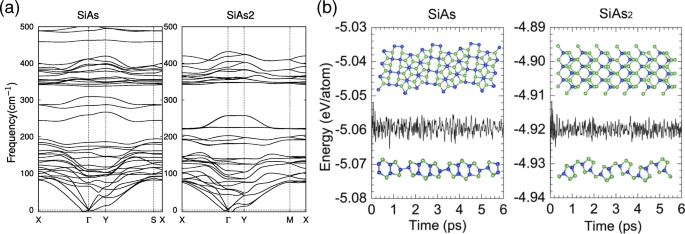
Curvas de dispersão de fônons e simulações de MD de monocamadas de SiAs e SiAs 2 . a As curvas de dispersão de fônons para SiAs e SiAs monocamada 2 . b Relações de energia total e tempo durante simulações de MD de SiAs e SiAs à temperatura ambiente 2 . Instantâneos selecionados das estruturas de monocamada no final de 6 ps também são fornecidos
Com as estruturas otimizadas de SiAs e SiAs monocamada 2 , agora prestamos atenção às suas propriedades eletrônicas. As estruturas de banda de decomposição orbital calculada de SiAs e SiAs 2 monocamadas são mostradas na Fig. 3. Nossos cálculos mostram claramente que SiAs e SiAs 2 monocamadas são semicondutores indiretos com grandes lacunas de banda. Para SiAs monocamada, a banda de valência máxima (VBM) está localizada em Y ponto, enquanto a banda de condução mínima (CBM) está no Γ (Fig. 3a). O gap indireto de SiAs monocamada é E g =1,72 eV no esquema PBE. Também se pode ver que o estado VBM em Y ponto compreende o p y orbital, enquanto o CBM de Γ O ponto compreende principalmente o orbital s, o que significa que a deformação externa terá efeitos diferentes nos dois estados e pode levar a uma transição indireta-direta, conforme revelado a seguir. Ao contrário de SiAs, a monocamada SiAs 2 é um semicondutor quase direto com VBM localizado ao lado do Y ponto e CBM está um pouco afastado dele (Fig. 3b). Os SiAs 2 a diferença de banda indireta da monocamada é E g =1,42 eV no esquema PBE. E o VBM e CBM de SiAs 2 monocamada são compostas de p y orbital es orbital s, respectivamente. A fim de obter um valor de gap mais preciso, também realizamos os cálculos funcionais híbridos (HSE06) [46, 47] para SiAs e SiAs 2 monocamadas. A partir das estruturas de banda calculadas (a parte direita da Fig. 3a, b), os sharps dos estados da banda de PBE e HSE são basicamente os mesmos, e o gap indireto ainda é previsto dentro dos cálculos funcionais híbridos, mas o valor do gap é aumentou para 2,39 eV e 2,07 eV para SiAs e SiAs 2 , respectivamente.
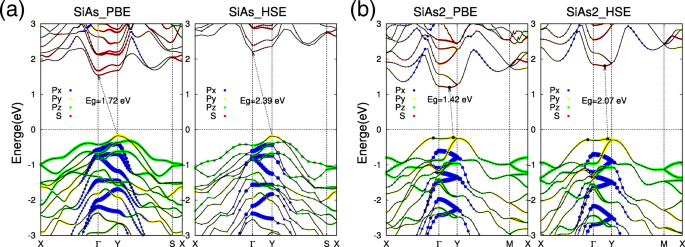
Estruturas de banda de SiAs e SiAs monocamada 2 calculado por PBE e HSE06. A decomposição orbital eletrônica de estruturas de banda de monocamadas SiAs e SiAs 2 são representados como um e b , respectivamente. Os pontos vermelhos denotam s orbital, enquanto azul, amarelo e verde são p x , p y e p z , respectivamente. O nível Fermi é definido como zero e indicado com uma linha pontilhada
As mobilidades do portador, que são um fator chave das aplicações potenciais em dispositivos eletrônicos modernos para os materiais 2D recém-descobertos, são tão importantes quanto o bandgap e a localização de CBM e VBM. Para obter mais detalhes sobre as propriedades da estrutura eletrônica de SiAs e SiAs 2 monocamadas, calculamos então suas mobilidades de portadoras limitadas por fônons acústicos (incluindo elétrons e lacunas nas direções xey) com base na teoria do potencial de deformação (DP) [48] à temperatura ambiente ( T =300 K ) No regime de baixa energia (300 K ), o espalhamento elétron-acústico-fônon domina o transporte da portadora, o que torna o fônon acústico limitado uma forma eficaz de prever as mobilidades da portadora de muitas estruturas 2D, como o MoS 2 monocamada [49], telureno [19], fosfeno [50] e MoO de poucas camadas 3 [51]. As massas efetivas calculadas m ∗ e mobilidades da portadora μ de SiAs e SiAs 2 monocamadas mostram que ambos são de alta mobilidade e anisotropia de transporte (ver arquivo adicional 1:Tabela S1 e as Figuras S3 e S4) como o fosforeno preto [50]. Para estimar a mobilidade do portador de SiAs e SiAs 2 , primeiro realizamos um ajuste de suas bandas usando o modelo de elétron quase livre para obter as massas portadoras efetivas. Para SiAs, definimos x e y como a direção perpendicular aos vetores da rede b e a , respectivamente. O \ (m_ {e} ^ {*} \) e \ (m_ {h} ^ {*} \) ao longo da direção x são cerca de 0,15 m 0 e 0,86 m 0 , respectivamente, e ao longo da direção y são 0,80 m 0 e 0,22 m 0 ( m 0 é a massa do elétron livre), respectivamente. Para SiAs 2 , a direção do vetor de rede a é definido como x , enquanto o de b é y . O \ (m_ {e} ^ {*} \) e \ (m_ {h} ^ {*} \) ao longo da direção x são cerca de 0,14 m 0 e 0,65 m 0 , respectivamente, e ao longo da direção y são 2,05 m 0 e 1,82 m 0 , respectivamente. Estudamos posteriormente as constantes elásticas (C) e os potenciais de deformação (E1) (consulte o arquivo adicional 1:Figura S2 e S3). Com base no acima obtido m ∗ , Valores C e E1, estimamos a mobilidade do portador conforme listado na Tabela 1. As mobilidades de elétrons para SiAs (SiAs 2 ) ao longo de x e y as direções são 0,66 (0,26) e 0,54 (0,11) × 10 3 · cm 2 V −1 S −1 , enquanto o buraco se mobiliza ao longo de x e y as direções são 3,90 (0,13) e 0,30 (0,65) × 10 3 · cm 2 V −1 S −1 , respectivamente, sendo que ambos são muito maiores do que os do MoS 2 [49].
Para esclarecer ainda mais o mecanismo de ligação subjacente dos átomos de Si e As em monocamadas SiAs e SiAs 2 , a densidade total e parcial de estados (PDOS) deles usando PBE funcional, com sua distribuição de densidade de elétrons correspondendo a VBM e CBM, são fornecidos na Fig. 4, respectivamente. Pode-se ver que o PDOS de átomos de As e Si (Fig. 4a, c) mostra uma forte hibridização de s e p orbitais, indicando a forte ligação covalente entre eles. As distinções entre monocamadas SiAs e SiAs 2 são a localização de p z orbitais, que são atribuídos ao ambiente de coordenação de ligação diferente do átomo de As. Os estados de elétron de par solitário, localizados no átomo de As em ambos os SiAs e SiAs 2 monocamadas, aumente os três orbitais de ligação mais próximos para decidir a formação de flambagem da estrutura da monocamada e para formar o p z ação de localização orbital. Em SiAs monocamada, os pares solitários são separados pela ligação Si – As, que relaxa o efeito repulsivo e amplia o p z orbital. Considerando que em monocamada SiAs 2 , Ligação As-As, permanecendo a situação que é muito comum em semicondutores do grupo V, localiza o p z orbital em um nível de energia mais profundo.
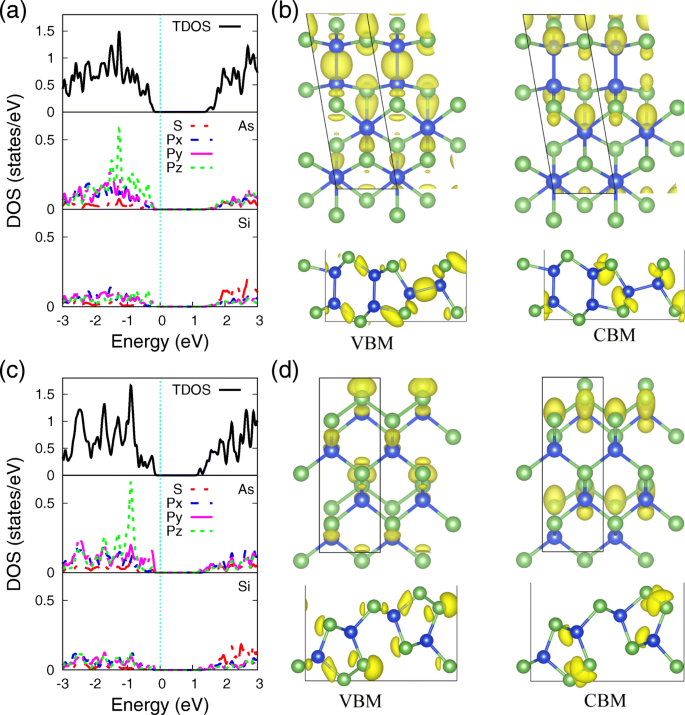
Densidade projetada de estados e densidade de elétrons de VBM e CBM. A densidade projetada de estados (PDOS) de átomos de As e Si e a distribuição de densidade de elétrons correspondente a VBM e CBM de ( a , b ) SiAs e ( c , d ) SiAs 2 monocamadas. O valor de isosuperfície 0,034 e / Å 3
Como sabemos, o caráter dos estados de fronteira não é apenas de interesse em uma compreensão microscópica dos canais de condução, mas também de grande preocupação para o projeto de contatos ideais. [52] As densidades de carga correspondentes a VBM e CBM de monocamadas SiAs e SiAs 2 são apresentados na Fig. 4b e d, respectivamente. O VBM é quase a hibridização de orbitais 3p de Si e As, enquanto CBM é principalmente da contribuição de orbitais 3s de Si e As, que também são consistentes com os resultados de PDOS na Fig. 4a, c e a decomposição orbital eletrônica de estruturas de banda na Fig. 3.
A deformação mecânica é uma forma eficaz de modular as propriedades eletrônicas de materiais 2D, que são amplamente usados para modificar a estrutura de banda de fosforenos pretos e azuis e outros materiais de nanofolhas [53-55]. Especialmente, para o sistema de estrutura curvada, o custo de energia é geralmente muito pequeno para induzir uma deformação acentuada. Aqui, a aplicação de deformação mecânica é simulada variando a constante de rede, bem como os graus de liberdade internos de cada átomo durante a otimização geométrica. A tensão ε é definido como ε =( l - l 0 ) / l 0 , onde l e l 0 são as constantes de rede tensas e de equilíbrio das monocamadas SiAs e SiAs 2 . Na Fig. 5a, b, as variações detalhadas da estrutura geométrica de alta flambagem de SiAs e SiAs 2D 2 sob as tensões são representadas, respectivamente. Pode-se ver que suas alturas de curvatura são expandidas ou comprimidas pela alteração do ângulo de curvatura θ 1 (2) com deformações compressivas ou de tração biaxiais em variações quase lineares. E também descobrimos que sua estrutura geométrica de alta flambagem ainda é mantida sob deformações bastante grandes, cujos espectros de fônons, como mostrado no Arquivo adicional 1:Figura S5 e S6, não existem frequências negativas mesmo no regime de deformação grande. As variações de intervalo de SiAs e SiAs monocamada 2 sob compressão biaxial e tensões de tração são mostradas na Fig. 5c, d, respectivamente. Pode-se ver que as propriedades eletrônicas de SiAs e SiAs 2 dependem sensivelmente da deformação e passam por uma transição de banda indireta para direta em certa região de deformação e, em seguida, para o metal em uma grande região de deformação.
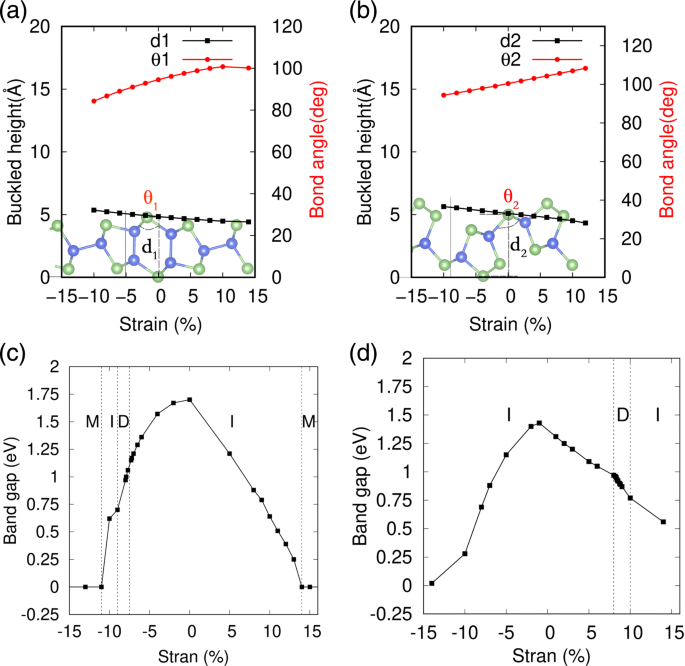
Efeitos de tensão nas estruturas geométricas e lacunas de banda de SiAs e SiAs 2D 2 . a , c representar SiAs; e b , d denotam SiAs 2 ; M, I e D representam metal, semicondutor indireto e semicondutor direto, respectivamente
As variações detalhadas de SiAs e SiAs 2 estruturas de banda são exibidas nas Figs. 6 e 7, respectivamente. Sob deformações compressivas biaxiais, a altura de curvatura da monocamada de SiAs está aumentando e o CBM muda de Γ para um ponto na linha Y – S e de volta para Y. Enquanto o VBM é mantido imóvel no ponto Y até que a deformação compressiva alcance ε =- 10 % . Portanto, com o aumento da deformação compressiva, o gap muda de Y indireto para Γ , via Y indireto para um ponto na linha Y – S, para direcionar Y para Y e de volta para um ponto indireto no Γ –Y linha para Y, como mostrado na Fig. 6. Para tensões de tração, o VBM em Y se move para um ponto na linha Y – S e o CBM em Γ move-se para Y e o gap permanece indireto. Para grandes deformações, não importa a compressão ou tração leva a uma transição de metal, como mostrado na Fig. 5c.
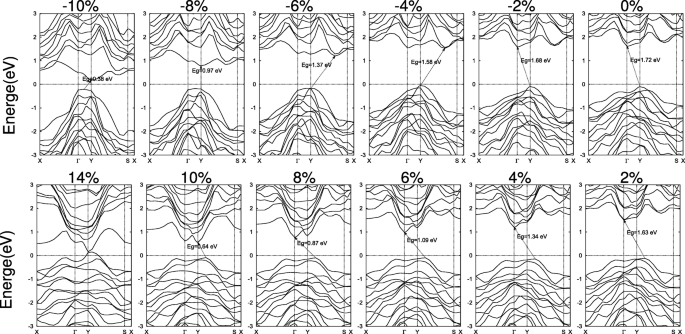
Estruturas de bandas de SiAs 2D sob as cepas biaxiais. O nível Fermi é definido como zero e indicado com uma linha pontilhada
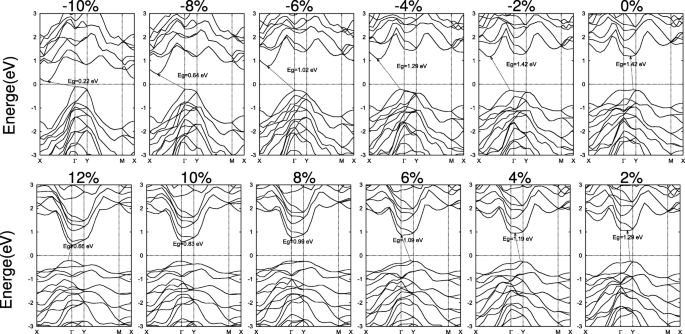
Estruturas de banda de SiAs 2D 2 sob as tensões biaxiais. O nível de Fermi é definido como zero e indicado com uma linha pontilhada
Na Fig. 7, um estudo semelhante foi realizado para 2D SiAs 2 . Em vez de compressão, as deformações de tração na faixa de 8 a 10% resultam em folgas de banda diretas. quando a monocamada SiAs 2 se espalha com uma diminuição da altura de dobra sob as deformações de tração, o VBM muda de um ponto no Γ –Y linha para Γ e mantenha-se imóvel na faixa de 8–10% e, em seguida, desloque-se para um ponto no Γ –X linha, enquanto o CBM se move de um ponto na Γ –Y linha para Γ e espere. Portanto, com o aumento da tensão de tração, o gap muda de indireto no Γ –Y linha para direcionar Γ - Γ e depois de volta para um ponto indireto no Γ –X linha para Γ , conforme ilustrado na Fig. 7. As deformações compressivas permanecem como o gap indireto. E grandes deformações têm efeitos semelhantes, levando a uma transição de metal como SiAs.
Estruturas de banda direta representativas de SiAs e SiAs tensos 2 também são mostrados no arquivo adicional 1:Figura S7a eb pelos cálculos PBE e HSE. Para SiAs, um gap direto de E g =1,75 eV (HSE) com o VBM e CBM localizados no Y pontos é obtido sob uma deformação compressiva biaxial de ε =- 7,5 % . Ao contrário de SiAs, uma deformação de tração biaxial de ε =8,5 % induz o SiAs 2 para uma banda direta de E g =1,60 eV (HSE). E o VBM e o CBM estão no Γ ponto.
Conclusões
Em resumo, realizando um cálculo DFT de primeiros princípios, propusemos dois novos tipos de materiais 2D de composto de silício e arsênio, SiAs e SiAs 2 , que são dinamicamente e termodinamicamente estáveis. Nossos cálculos mostram que SiAs e SiAs 2 monocamadas são semicondutores indiretos com lacunas de banda de 2,39 eV e 2.07 eV , respectivamente. O intervalo de banda de SiAs e SiAs 2 as monocamadas são sensíveis à deformação, que passa por uma transição de banda indireta para direta e até mesmo para o metal após certa deformação mecânica. SiAs e SiAs 2 monocamadas possuem maior mobilidade do que MoS 2 e exibir transporte anisotrópico como o fosforeno preto. Nossos trabalhos abrem um novo caminho em nanoescala para novas funcionalidades de dispositivos ópticos.
Abreviações
- 2D:
-
Bidimensional
- CASTEP:
-
Pacote de energia total sequencial de Cambridge
- CBM:
-
Banda de condução mínima
- DFT:
-
Teoria da densidade funcional
- DFPT:
-
Teoria de perturbação funcional de densidade
- DP:
-
Potencial de deformação
- GGA:
-
Aproximação de gradiente generalizado
- MD:
-
Dinâmica Molecular
- NVT:
-
Moles-volume-temperatura
- PAW:
-
Onda aumentada do projetor
- PBE:
-
Perdew-Burke-Ernzerhof
- PDOS:
-
Densidade parcial de estados
- TMDs:
-
Dichalcogenetos de metal de trânsito
- VASP:
-
Pacote de simulação de Viena ab initio
- VBM:
-
Banda de valência máxima
Investigação da Faixa de Energia nas Heterojunções de Bissulfeto de Molibdênio e ZrO2
Análise comparativa de defeitos em camadas de GaN implantadas e dopadas com magnésio em substratos de GaN independentes
Nanomateriais
- Remoção adsortiva de íons de cobre (II) de solução aquosa usando um nanoadsorvente de magnetita de resíduos de escala de moinho:Síntese, caracterização, adsorção e modelagem cinética Estud…
- Absorvedor perfeito de banda ultra-estreita e sua aplicação como sensor plasmônico na região visível
- Estudo de primeiros princípios sobre a estabilidade e imagem STM de Borophene
- RGO e redes de grafeno tridimensionais TIMs co-modificados com alto desempenho
- Preparação de nanoesferas poliméricas impressas com íons de paládio (II) e sua remoção de paládio (II) da solução aquosa
- Nanomontagens de ácido 5-aminolevulínico-esqualeno para fotodetecção e terapia de tumor:estudos in vitro
- Estudo dos primeiros princípios de defeitos pontuais na superrede de GaAs / AlAs:a estabilidade de fase e os efeitos na estrutura da banda e na mobilidade do portador
- Propriedades eletrônicas ajustáveis por deformação e alinhamentos de banda na heteroestrutura GaTe / C2N:um cálculo dos primeiros princípios
- Investigação da Faixa de Energia nas Heterojunções de Bissulfeto de Molibdênio e ZrO2
- Strain Tunable Bandgap e High Carrier Mobility em SiAs e SiAs2 Monocamadas de Estudos de Primeiros Princípios