


Estudo dos primeiros princípios de penta-siligrafeno como material de ânodo de alto desempenho para baterias de íons de lítio
Resumo
A partir dos cálculos dos primeiros princípios, prevê-se que uma nova complexidade pentagonal de Si / C tenha aplicações potenciais como um material de ânodo promissor para baterias de íons de lítio. Verificou-se que a estabilidade estrutural e térmica do penta-siligrafeno (P-Si 2 C 4 ) é melhor do que o pentagrafeno, composto apenas de átomos de carbono. A análise da estrutura de banda eletrônica mostra que o C-2 vazio p z estado no P-Si 2 C 4 fornece espaço para acomodar e estabilizar os elétrons de Li, o que torna o armazenamento de Li energeticamente favorável. Como resultado, quatro átomos de Li podem ser armazenados por uma unidade de fórmula do P-Si 2 C 4 , correspondendo a uma capacidade teórica de armazenamento gravimétrico de Li de 1028,7 mAhg −1 . As estruturas eletrônicas metálicas do P-Li adsorvido a lítio x Si 2 C 4 bem como barreiras de energia de migração de Li muito pequenas são benéficas para o desempenho de carga / descarga rápida da bateria. O mecanismo na interação de adsorção de Li no P-Si 2 C 4 é discutido. Esses resultados demonstram uma nova estratégia para projetar materiais anódicos complexos de Si / C bidimensionais para baterias de íon-lítio de alto desempenho.
Histórico
A densidade de energia relativamente baixa das baterias de íon-lítio (LIBs) comercializadas atualmente é difícil de atender aos requisitos dos veículos elétricos comerciais (EVs) e se torna um grande desafio para o desenvolvimento da indústria de EVs [1, 2]. Para aumentar a densidade de energia dos LIBs, precisamos melhorar a capacidade dos materiais do eletrodo. Devido ao seu desempenho de ciclismo muito bom, o grafite é o material anódico mais amplamente usado, mas sua capacidade gravimétrica teórica (372 mAhg −1 ) é relativamente baixo [3, 4]. Por outro lado, o silício tem capacidade gravimétrica teórica extremamente alta de cerca de 4200 mAhg −1 [5], mas o desempenho do ciclismo é ruim devido à sua grande expansão de volume de até 420% no estado totalmente litiado [6]. Para aproveitar as vantagens dos ânodos de silício e de carbono, projetar um ânodo complexo de Si / C é acadêmico e tecnologicamente significativo.
O siligrafeno, que é um material em camadas semelhante ao grafeno bidimensional (2D) com átomos de C parcialmente substituídos por átomos de Si, foi inicialmente previsto para ser um material 2D estável a partir de cálculos de primeiros princípios [7,8,9,10] e tinha sido preparado com sucesso a partir de experimentos [11, 12]. Lin et al. mostraram que folhas de SiC 2D podem ser preparadas por meio de técnicas de esfoliação em solução [11]. Eles também prepararam com sucesso SiC quase 2D 2 folhas que podem ser preservadas ao ar por meses [12]. Mais tarde, os cálculos dos primeiros princípios sugerem que o siligrafeno é um material anódico promissor que oferece uma capacidade teórica de 1520 mAhg −1 e 1286 mAhg −1 para g-SiC 5 e g-SiC 2 , respectivamente [13]. É mostrado que o ânodo de siligrafeno herda a alta estabilidade de ciclo dos ânodos de grafite, bem como a alta capacidade dos ânodos de silício. A alta capacidade de armazenamento de Li prevista é atribuída à interação de adsorção de Li aprimorada com a monocamada de siligrafeno, que está relacionada às mudanças do átomo de Si de sp 2 para sp 3 -como [13]. No entanto, a configuração eletrônica mudou de sp 2 para sp 3 como é acompanhado por mudanças estruturais óbvias durante a adsorção de Li no siligrafeno. Isso não é bom para o desempenho cíclico do siligrafeno como um material anódico para LIBs. A melhor solução é projetar materiais complexos de Si / C que já possuem sp 3 -como configuração eletrônica.
O carbono tem muitos tipos de alótropos, que são formados com sp , sp 2 e sp 3 hibridização ou suas combinações. O muito estável sp 2 mais a configuração eletrônica de grande ligação π no carbono grafite é responsável pelas fracas interações de adsorção de Li no grafeno. Após a adsorção de Li em grafeno monocamada, a transferência de carga ocorre de Li para a camada de grafeno [14]. Então, Li torna-se positivamente carregado e se liga à camada de grafeno pela interação atrativa de Coulomb. No entanto, o excesso de carga de Li na camada de grafeno quebra a grande ligação π do grafeno, que é energeticamente desfavorável. Como resultado, a adsorção de Li no grafeno de camada única não é favorecida com energia de adsorção negativamente maior do que a energia coesiva da face centrada no corpo ( bcc ) Fase Li metal, o que não é permitido em baterias de íon-lítio. Como resultado, o armazenamento de Li com monocamada de grafeno puro não é permitido [15]. Alternativamente, os materiais de carbono duro oferecem uma capacidade gravimétrica de armazenamento de Li / Na muito maior em comparação com os materiais de carbono grafite [16,17,18]. O material de carbono duro é conhecido como uma fase amorfa que é composta por sp 2 e sp 3 átomos de carbono [19]. É possível que a maior capacidade gravimétrica de armazenamento de Li de materiais de carbono duro esteja relacionada ao sp 3 configuração eletronica?
Penta-grafeno, que é conhecido como um sp 2 - sp 3 alótropo de carbono 2D híbrido [20], foi previsto como um material anódico promissor para baterias de íons de lítio / Na a partir dos cálculos dos primeiros princípios [21]. Como um alótropo de carbono 2D, o penta-grafeno tem um comportamento de adsorção de Li muito mais forte em comparação com o grafeno convencional com estrutura em favo de mel. Este comportamento diferente de adsorção de Li também está relacionado ao sp 3 -como configuração eletrônica no penta-grafeno? Se a resposta for sim, qual é o mecanismo intrínseco por trás disso?
Embora seja previsto que o pentagrafeno seja um alótropo de carbono dinamicamente estável, sua energia coesiva é significativamente maior em comparação com a fase globalmente mais estável (grafite ou grafeno). A energia coesiva do pentagrafeno é cerca de 0,9 eV por átomo maior do que a do grafeno hexagonal de camada única [20, 22], o que torna muito difícil (se possível) a fabricação em larga escala do pentagrafeno industrialmente. No entanto, quanto às aplicações como material de ânodo, a fabricação em larga escala é muito importante. Observe que a flambagem é encontrada no siliceno e, portanto, o Si é mais estável com sp 3 como hibridização do que sp 2 [23,24,25] enquanto átomos de C preferem sp 2 hibridização em estruturas 2D; é razoável especular que substituir o sp 3 como átomos de C com átomos de Si na estrutura do pentagrafeno serão energeticamente favorecidos. Chamamos essa estrutura de pentassiligrafeno. Experimentos recentes demonstraram que nanofitas à base de Si pentagonais podem ser cultivadas em Ag (110) [26], mostrando que a formação de estruturas pentagonais à base de Si é experimentalmente possível.
Teoricamente, a natureza eletrônica e de ligação do pentassiligrafeno (P-SiC 2 ) foi estudado por Lopez-Bezanilla et al. e eles descobriram que P-SiC 2 exibe uma inversão parcial da ordenação vertical das bandas eletrônicas p-p-σ e p-p-π [27]. Posteriormente, as propriedades de transporte eletrônico do P-SiC 2 são estudados e comparados com penta-grafeno e penta-CN 2 [28]. Curiosamente, é demonstrado que o desempenho de transporte eletrônico do P-SiC 2 pode ser ajustado por meio de engenharia de deformação, e foi previsto que a deformação compressiva uniaxial é capaz de aumentar a mobilidade do orifício da monocamada penta-SiC 2 até 1,14 × 10 6 cm 2 V −1 s −1 [29]. Apesar da semelhança nas estruturas, o pentassiligrafeno tem propriedades de transporte diferentes em comparação com o pentassiligrafeno. Foi encontrado por Hu et al. que a condutividade térmica do penta-grafeno exibe redução monotônica padrão por alongamento, enquanto o penta-SiC 2 possui um comportamento incomum não monotônico para cima e para baixo [30]. Essas propriedades interessantes do pentassiligrafeno estão fortemente relacionadas à natureza eletrônica e química dos átomos de Si na estrutura. Também foi descoberto que o próprio elemento de Si é benéfico para aumentar a adsorção de Li, uma vez que a interação de adsorção de Li no siliceno é muito mais forte em comparação com a do grafeno [31, 32]. Portanto, pode ser interessante saber se o pentassiligrafeno pode ser usado como materiais anódicos para LIBs.
Neste trabalho, investigamos os comportamentos de armazenamento de íons de lítio no penta-siligrafeno com cálculos de primeiros princípios, e o mecanismo de como o íon de lítio pode ser armazenado pelo penta-siligrafeno é discutido especificamente. Iniciamos nosso estudo a partir da estabilidade termodinâmica do penta-siligrafeno, seguido de uma análise detalhada das interações intrínsecas de adsorção de Li nele. Finalmente, o desempenho do pentassiligrafeno como um material anódico para LIBs é discutido.
Métodos Computacionais
Todos os cálculos neste trabalho são realizados usando o Vienna Ab initio Simulation Package (VASP) [33] com base na teoria do funcional da densidade (DFT). O método de onda aumentada do projetor (PAW) [34, 35] combinado com os funcionais de troca e correlação de aproximação de gradiente geral (GGA) parametrizados por Perdew-Burke-Ernzerhof (PBE) são usados [36]. A energia de corte para as ondas planas é escolhida como sendo 450 eV para todos os cálculos. Os parâmetros de rede e as posições iônicas são totalmente relaxados e as forças finais são convergidas para 0,02 eV / Å. A estrutura da banda eletrônica é calculada com o funcional híbrido Heyd-Scuseria-Erznerhof (HSE06) [37], pois o funcional híbrido tem uma descrição mais precisa da estrutura eletrônica. O cálculo da densidade de estados (DOS) é borrado pelo método de esfregaço gaussiano com largura de esfregaço de 0,05 eV. O Monkhorst-Pack [38] k - amostragem de pontos é usada e a densidade do k -mesh é mais espesso que 0,05 Å −1 para simulação de dinâmica de molécula ab initio (AIMD) e 0,03 Å −1 para outros cálculos. A distribuição de carga atômica é analisada com a análise de carga de Bader [39]. A via de migração de íons de lítio é otimizada com o método de banda elástica nudged imagem escalada (CINEB) [40]. A energia de adsorção E anúncio é calculado por:
$$ {E} _ {\ mathrm {ad}} =\ left ({E} _ {\ mathrm {host} + n \ mathrm {Li}} - {E} _ {\ mathrm {host}} - {nE } _ {\ mathrm {Li}} \ right) / n $$
onde E host , E Li , e E host + Li são as energias totais dos materiais hospedeiros penta-siligrafeno, o átomo de Li e os hospedeiros adsorvidos por Li, respectivamente, n denota o número de íons Li adsorvidos no penta-siligrafeno. A influência das interações de van der Waals (vdW) na energia de adsorção é testada usando o método DFT-D3 com amortecimento de Becke-Jonson [41]. Além da energia de adsorção, o potencial médio de intercalação de Li (vs Li + / Li) pode ser obtido diretamente da diferença da energia de adsorção e da energia coesiva do metal Li (fase bcc) de V ave =- ( E anúncio - E Li - coeso ), se escolhermos eV e V como as unidades de energia e potencial, respectivamente.
Resultados e discussões
Estrutura e estabilidade do penta-siligrafeno
A estrutura do pentagrafeno (ver Fig. 1a, denotada como P-C 6 no seguinte deste artigo) possui o P-42 1 m simetria (grupo espacial nº 113). As constantes de rede otimizadas são a = b =3,636 Å, de acordo com resultados anteriores [20, 21]. Dois tipos de átomos de carbono podem ser encontrados na estrutura, nomeadamente, carbono coordenado em 4 (denotado como C1 na Fig. 1a) e carbono coordenado em 3 (denotado como C2 na Fig. 1a). Pela geometria local dos átomos de carbono, podemos ver que C1 é sp 3 -como hibridizado enquanto C2 é sp 2 -como hibridizado. Embora o átomo C2 seja considerado sp 2 como hibridizado [20], o recurso de ligação dupla C2-C2 torna o caráter químico do átomo C2 diferente daquele do grafeno, o que será discutido em detalhes a seguir neste artigo. Substituindo átomos C1 por átomos de Si no P-C 6 estrutura, o penta-siligrafeno é formado (ver Fig. 1b, o arquivo de informação cristalina da estrutura otimizada é dado no arquivo adicional 1:SI-1 do material suplementar) e denotado como P-Si 2 C 4 no seguimento deste artigo. Como o raio atômico do átomo de Si é maior do que o do átomo C, as constantes de rede do P-Si 2 C 4 ( a = b =4,405 Å) é maior do que P-C 6 , enquanto em boa concordância com os outros resultados relatados [27,28,29,30].
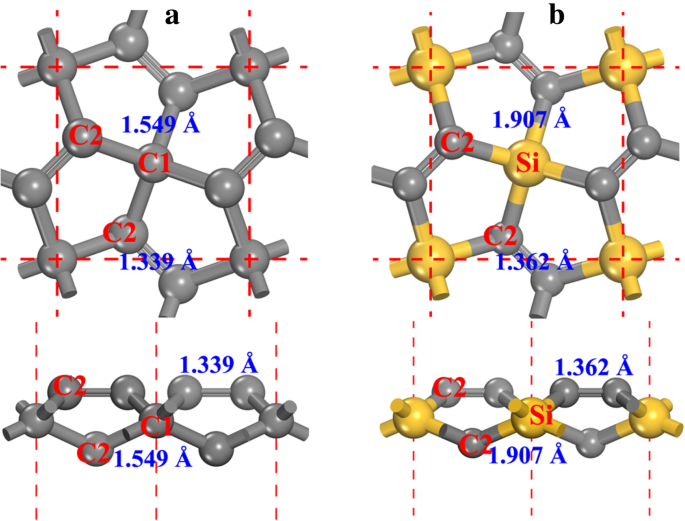
a O modelo bola e bastão do pentagrafeno e b penta-siligrafeno. Ambas as vistas de topo (para cima) e laterais (para baixo) são apresentadas. As esferas cinza e amarela são átomos C e Si, respectivamente. Os átomos de carbono coordenados de 4 e 3 são denotados como C1 e C2, respectivamente. Os comprimentos da ligação também são apresentados ao lado de cada ligação
Para avaliar a estabilidade termodinâmica relativa, a Tabela 1 apresenta as energias coesivas de diferentes alótropos dos complexos C, Si e C / Si. Embora penta-grafeno (P-C 6 ) é mostrado para ser estável a 1000 K a partir de simulações de dinâmica molecular ab initio (AIMD) [20], a energia coesiva de P-C 6 (- 8,24 eV · átomo −1 ) é muito maior em comparação com o grafeno de camada única (- 9,14 eV · átomo −1 ) Isso mostra que a produção em massa de P-C 6 deve ser muito difícil. Por outro lado, a energia coesiva de P-Si 2 C 4 (- 7,26 eV · átomo −1 ) é apenas 0,2 eV mais alto em comparação com seu alótropo mais estável g-Si 2 C 4 (- 7,46 eV · átomo −1 ), mostrando que a preparação de pentassiligrafeno pode ser muito mais fácil em comparação com P-C 6 . Para verificar a estabilidade estrutural do P-Si 2 C 4 , curvas de dispersão de fônons do P-Si 2 C 4 foram calculados e apresentados na Fig. 2. Embora pequenas frequências imaginárias sejam encontradas em uma pequena região perto do ponto Γ (0,0039 THz ou 0,13 cm −1 ), ainda podemos acreditar que o sistema é dinamicamente estável, porque é geralmente aceito que essas pequenas frequências imaginárias (não maiores que 1 cm −1 ) poderia ser um artefato da simulação [42]. Frequências imaginárias também foram relatadas em outros materiais 2D dinamicamente estáveis, como germaneno [43] e arseneno [44]. Aplicando tratamentos técnicos como aumentar a precisão do cálculo ou usar métodos de cálculo diferentes, essas frequências imaginárias podem ser removidas.
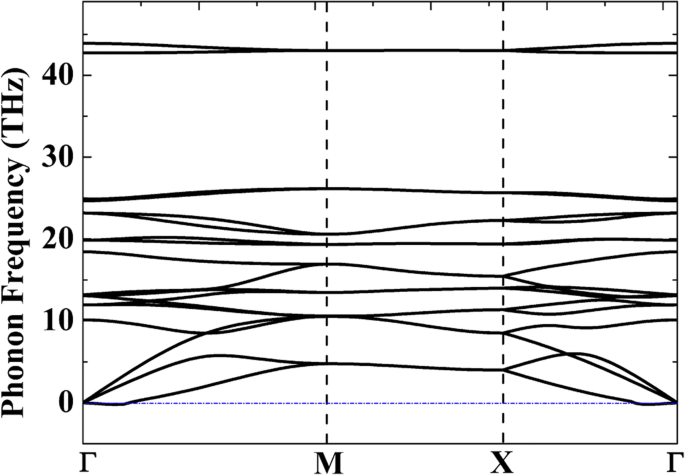
Curvas de dispersão de fônons do 2D P-Si 2 C 4 monocamada calculada a partir da teoria da resposta linear
Além disso, a simulação AIMD também é realizada para avaliar a estabilidade estrutural de P-Si 2 C 4 em altas temperaturas. AIMD são realizados usando supercélulas 3 × 3 e 4 × 4 em um conjunto canônico a temperaturas de 1000 K, 1500 K, 2000 K e 2500 K (consulte o arquivo adicional 1:Figura S1). Arquivo adicional 1:As Figuras S2 e S3 apresentam as configurações atômicas do P-Si 2 C 4 no final das simulações AIMD em diferentes temperaturas usando supercélulas 3 × 3 e 4 × 4, respectivamente. Como é mostrado, os anéis atômicos pentagonais são mantidos inalterados quando a temperatura é tão alta quanto 2.000 K durante o tempo de simulação de 20 ps, mostrando que a estrutura pode suportar uma temperatura tão alta quanto 2.000 K. Por outro lado, graves deformações estruturais são observados e anéis hexagonais (ver arquivo adicional 1:Figura S2d), bem como outros defeitos (arquivo adicional 1:Figura S3d) apareceram nos instantâneos, indicando que as estruturas são destruídas em 2500 K. Os anéis hexagonais encontrados em P-Si 2 C 4 a 2500 K indicam que g-Si 2 C 4 (que é composto de anéis hexagonais [13]) é mais estável do que a fase-penta-penta 2 C 4 , consistente com a energia coesiva dada na Tabela 1. Estes resultados confirmam que a estabilidade estrutural do P-Si 2 C 4 é muito mais estável em comparação com o do P-C 6 , que só pode suportar uma temperatura de 1000 K.
Adsorção de Li em Penta-siligrafeno
Para estudar a adsorção de Li no penta-siligrafeno P-Si 2 C 4 , diferentes locais de adsorção de Li são considerados e quatro locais de adsorção estáveis (como mostrado na Fig. 3a) podem ser encontrados após o relaxamento. Os locais estáveis de adsorção de Li são o local superior do átomo de Si (denotado como T), o local oco (denominado H) do Si 2 C 3 anel pentágono e locais de ponte entre dois átomos C2 na camada inferior (B1) e na camada superior (B2). A preferência de adsorção de íons de lítio nestes sites pode ser caracterizada pelas energias de adsorção apresentadas na Tabela 2. Os resultados mostram que o sítio de adsorção de Li mais estável é o sítio B1, com energia de adsorção de -1,922 eV. Por outro lado, a energia de adsorção no local H (-1,905 eV) está muito próxima do local B1. As energias de adsorção de Li também são representadas pelas alturas de adsorção, pois a menor energia de adsorção corresponde a menores alturas de adsorção (ver Tabela 2). No início do processo de adsorção de Li, os íons de Li são preferidos para permanecer nos locais B1 mais estáveis. Depois que todos os locais B1 estiverem ocupados (correspondendo a uma estequiometria de Li 2 Si 2 C 4 e veja a Fig. 3b), os íons Li começam a permanecer em locais H. Como a distância entre os locais B1 e H é muito pequena (~ 1,5 Å), ocorre forte interação de repulsão para os íons Li nos locais B1 e H. Como resultado, os íons de Li nos locais B1 são repelidos para os locais H próximos e, portanto, os locais B1 ficam vazios enquanto todos os locais H estão ocupados no estado de Li 4 Si 2 C 4 (ver Fig. 3c). A influência da interação vdW para a energia de adsorção de Li também é testada, e os resultados são dados entre parênteses na Tabela 2. Como é mostrado, a interação vdW contribui de - 0,12 a - 0,17 eV para a energia de adsorção para diferentes locais de adsorção, mostrando que a interação vdW é a favor da adsorção de Li.
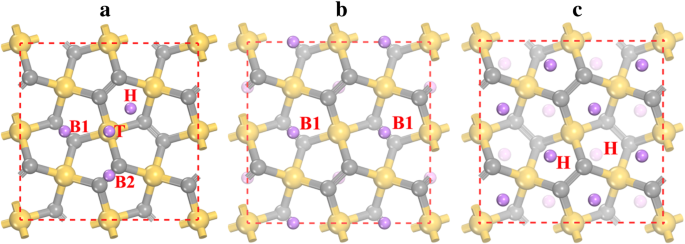
Sites de adsorção de Li em a superfície do penta-siligrafeno e a configuração atômica das estruturas mais estáveis de b Li 2 Si 2 C 4 e c Li 4 Si 2 C 4 . As esferas amarela (maior), cinza (tamanho médio) e roxa (menor) são átomos de Si, C e Li, respectivamente. H, T, B1 e B2 denotam os locais de adsorção de Li
Comparando as energias de adsorção de Li com a energia coesiva do bcc fase Li metal (-1,86 eV · átomo −1 ), podemos julgar se a adsorção de Li é eletroquimicamente ativa ou não. Se a energia de adsorção for inferior à energia coesiva do metal Li, a adsorção de íons de lítio é favorecida e a adsorção corresponde a um potencial de descarga positivo. Conforme apresentado na Tabela 1, as energias de adsorção em ambos os locais B1 e H são inferiores a -1,86 eV, mostrando que ambos os locais B1 e H são locais eletroquimicamente ativos para o armazenamento de Li. Para avaliar a capacidade de armazenamento de Li, as energias de adsorção de Li em diferentes concentrações de íons de lítio x (Razão Li / (Si + C)) são calculados e comparados com a energia coesiva de bcc Li metal. Como é mostrado na Fig. 4, as energias de adsorção de Li são menores que -1,86 eV quando a relação Li / C x é menor que 2/3, correspondendo a 4 átomos de Li adsorvidos em uma célula unitária do P-Si 2 C 4 e uma capacidade gravimétrica teórica de 1028,7 mAhg −1 (Li 4 Si 2 C 4 ) A densidade de energia de uma bateria, que é igual à capacidade vezes a tensão de saída, está mais preocupada em comparação com a capacidade de armazenamento de Li. Um bom material de ânodo deve ter potenciais eletroquímicos relativamente baixos, que podem ser obtidos a partir das energias de adsorção. O potencial médio é de cerca de 0,1–0,2 V, que é relativamente baixo e benéfico para uma tensão de saída mais alta de um sistema de bateria completo. Além disso, a Fig. 4 também apresenta as mudanças constantes da rede em diferentes concentrações de adsorção de Li. Como é mostrado, a constante de rede do P-Si 2 C 4 encolhe ligeiramente com a adsorção de Li. Quando a concentração x for 1/6, a mudança de rede atinge o maior valor, que é tão pequeno quanto - 0,94%, indicando que a mudança de volume será muito pequena durante o processo de carga / descarga. A pequena mudança de volume é benéfica para manter a estrutura do P-Si 2 C 4 para ser estável durante o ciclismo.
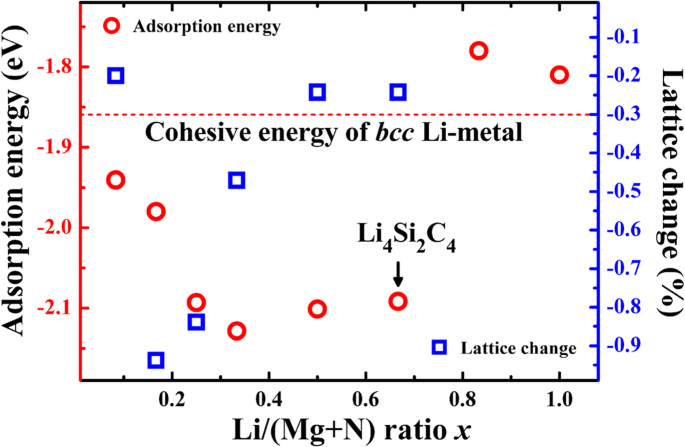
As energias de adsorção de Li calculadas em P-Si 2 C 4 superfície (ciclos vermelhos, eixo do lado esquerdo) e a mudança constante da rede (quadrados azuis, eixo do lado direito) em função da concentração de adsorção de Li (razão Li / (C + Si)). A energia coesiva de bcc fase Li metal também está incluído para comparação
Análise da Estrutura Eletrônica do Penta-siligrafeno após Adsorção de Li
As estruturas de banda eletrônica do penta-siligrafeno (P-Si 2 C 4 ) e seus estados litiados são apresentados na Fig. 5. Como pode ser visto, P-Si 2 C 4 é um semicondutor com um gap indireto de cerca de 2,35 eV, que é muito menor em comparação com 3,46 eV do pentagrafeno P-C 6 (consulte o arquivo adicional 1:Figura S4). O menor gap de P-Si 2 C 4 do que P-C 6 é originado da dispersão intensificada das bandas ocupadas mais altas (Nos. 11 e 12 na Fig. 5a), particularmente nos pontos de alta simetria M e Γ. O gap é aberto entre as bandas de energia do nº 12 (a banda ocupada mais alta) e o nº 13 (a banda desocupada mais baixa). O nível de energia da banda nº 12 aumenta substancialmente no ponto M, o que aumenta o nível de Fermi e, por sua vez, diminui o gap.
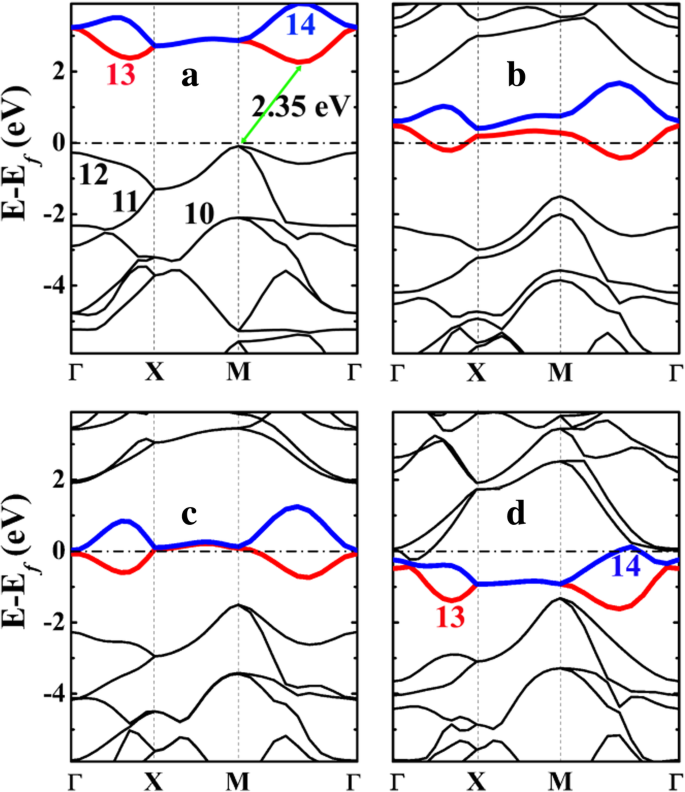
As estruturas de banda eletrônica de a penta-siligrafeno P-Si 2 C 4 e seus estados litiados b P-LiSi 2 C 4 , c P-Li 2 Si 2 C 4 , e d P-Li 4 Si 2 C 4 calculado a partir de HSE06. O nível de Fermi é selecionado para ser 0 eV. Os números de 10 a 14 em a e d denotam o número da banda, enquanto os números da banda 13 e 14 são destacados com as cores vermelha e azul, respectivamente. A rotulagem de bandas está em conformidade com o código VASP, no qual a rotulagem de banda é referenciada às bandas de valência e condução e os elétrons centrais não são incluídos na rotulagem
A partir da análise da forma da densidade de carga (função de onda) projetada para as bandas nº 10-14 mostradas na Fig. 6, podemos ver que a banda nº 12 corresponde aos estados de ligação das ligações σ formadas entre C e Si átomos, enquanto a banda No. 13 (e 14) corresponde ao 2- p z estado do átomo C. O vazio C- p z O estado fornece espaço para acomodar e estabilizar os elétrons da adsorção de Li, o que torna o processo de adsorção de Li energeticamente favorável.
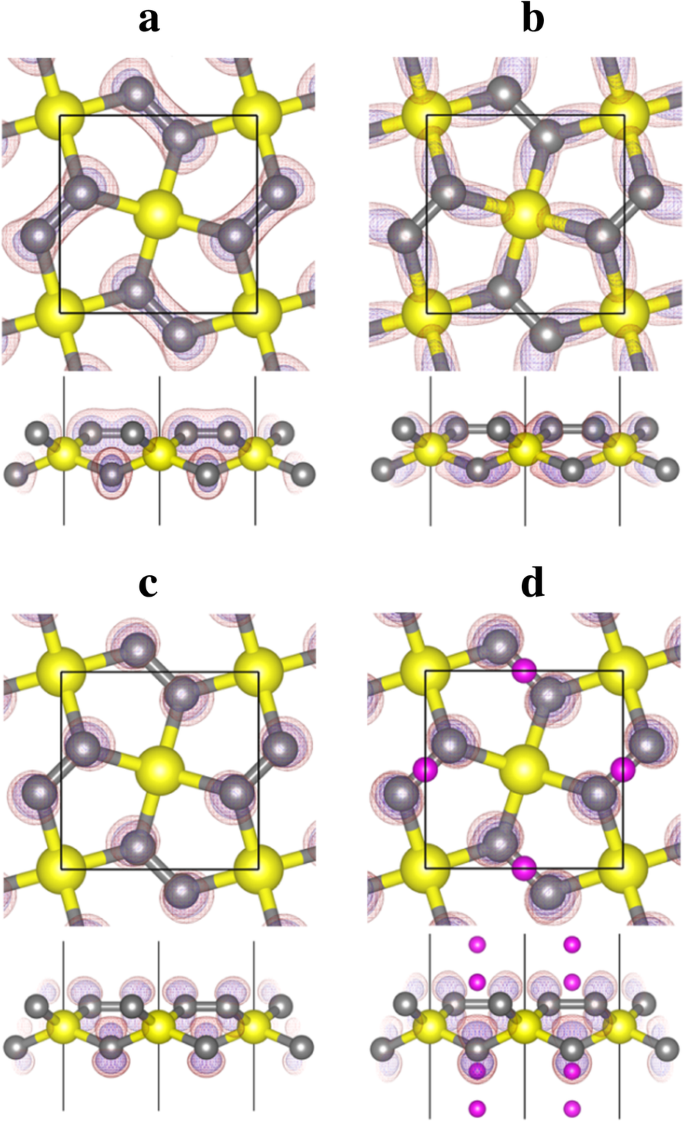
Contornos de densidade de carga decomposta em banda para bandas a No. 10, b No. 12, e c No. 13 do penta-siligrafeno (P-Si 2 C 4 , Fig. 5a) e banda d No. 13 do penta-siligrafeno litiado (Li 4 Si 2 C 4 , Fig. 5d). As esferas amarela (grande), cinza (tamanho médio) e roxa (pequena) são átomos de Si, C e Li, respectivamente. Os contornos da densidade de carga são exibidos em vermelho transparente (com valor de isosuperfície de 0,02 e / Å 3 ) e azul (com valor de isosuperfície de 0,01 e / Å 3 ) cores
A dispersão aprimorada das bandas de energia ocupadas mais altas em P-Si 2 C 4 pode ser atribuída a dois fatores:em primeiro lugar, a atração de Coulomb entre os elétrons que ocupam a banda nº 12 (ligações σ C – Si) e o átomo de Si carregado positivamente, em comparação com aquele no PC de penta-grafeno somente carbono 6 . A análise de carga de Bader mostra que o átomo de Si está positivamente carregado no penta-siligrafeno P-Si 2 C 4 . Conforme mostrado na Tabela 3, a carga de Bader de átomos de Si em siligrafeno ou penta-siligrafeno é de cerca de 1,65 e , mostrando que os átomos de Si são carregados positivamente com + 2,35 e . Por outro lado, o átomo C1 no penta-grafeno (P-C 6 ) também tem carga positiva, mas a carga líquida é de apenas + 0,08 e . Portanto, além da interação de ligação covalente, fortes interações de Coulomb entre átomos de C e Si ocorrem em P-Si 2 C 4 , em comparação com o do P-C 6 . Isso é benéfico para a dispersão das bandas de energia ocupadas (nº 12) perto do nível de Fermi. Em segundo lugar, a flambagem aprimorada no P-Si 2 C 4 também pode contribuir para a dispersão da banda nº 12 (o estado de ligação da ligação σ C – Si), pois uma flambagem maior denota mais sp 3 -hibridizadas e ligações σ mais fortes são formadas entre os átomos C – Si. Esta também é uma razão importante para a estabilidade estrutural estável do P-Si 2 C 4 em comparação com o de P-C 6 . Mais importante ainda, a flambagem entre os átomos de Si e C aumenta com a adsorção de Li e se torna 0,876 Å no estado litiado P-Li 4 Si 2 C 4 . Neste estado, os ângulos de ligação C – Si – C (102,50 ° e 124,59 °) do SiC 4 tetraedro torna-se mais próximo de 108,47 ° de um tetraedro padrão, mostrando que o sp 3 -hibridização do átomo de Si e a força das ligações C – Si tornam-se mais fortes após a adsorção de Li. Consequentemente, a dispersão da banda No. 12 também é aumentada com o aumento da concentração de adsorção de Li, como pode ser visto na Fig. 4.
Após a adsorção de Li na superfície do penta-siligrafeno (P-Si 2 C 4 ), transferências de carga dos átomos de Li (em operação real com bateria, Li + íons vêm do circuito interno, enquanto a mesma quantidade de elétrons vêm do circuito externo) para os átomos de carbono em P-Si 2 C 4 . Como resultado, o excesso de elétrons se moverá para baixo nas bandas desocupadas (bandas nº 13 e 14) dando origem às estruturas eletrônicas metálicas do sistema, conforme mostrado na Fig. 6b-d. As estruturas eletrônicas metálicas garantem uma boa condutividade eletrônica do P-Si 2 C 4 ânodo durante o processo de carga / descarga, o que é benéfico para o desempenho da taxa do sistema de bateria usando o P-Si 2 C 4 ânodo.
Como discutido acima e mostrado pela densidade de carga projetada da banda na Fig. 6, as bandas No. 13 e 14 são p z estados do átomo C em P-Si 2 C 4 . Essas bandas vazias são muito importantes para a adsorção de Li. Além da atração de Coulomb entre os íons Li carregados positivamente e os P-Si carregados negativamente 2 C 4 substrato, os elétrons ocupando o C- p z os estados têm uma forte atração de Coulomb para o átomo de Si carregado positivamente (que desce os níveis de energia das bandas nº 13 e 14 e, portanto, reduz a energia total do substrato). Como consequência, a adsorção de Li em P-Si 2 C 4 é energeticamente mais favorável. Como uma célula unitária do P-Si 2 C 4 contém 4 átomos de C, espera-se que 4 átomos de Li possam ser adsorvidos no P-Si 2 C 4 superfície. Após o C- p z estados estão completamente ocupados, adsorção de mais átomos de Li no P-Si 2 C 4 será energeticamente desfavorável. Isso está de acordo com as energias de adsorção calculadas apresentadas na Fig. 4.
Dinâmica de migração de íons de lítio em P-Si 2 C 4
A taxa de desempenho do P-Si 2 C 4 o ânodo é determinado por condução eletrônica e dinâmica de difusão de íons de lítio. Conforme discutido acima, embora a estrutura eletrônica do prístino P-Si 2 C 4 é um isolante, ele se torna metálico espontaneamente após a adsorção de Li, mesmo quando a concentração de Li é baixa. Portanto, a condutividade eletrônica deve ser boa o suficiente para aplicação como um material de ânodo. Então, a difusão de íons de lítio no penta-siligrafeno torna-se a etapa de controle de taxa. Como a estrutura do P-Si 2 C 6 é semelhante ao do pentagrafeno (P-C 6 ) no qual o íon de lítio se difunde muito rápido [21], espera-se que a difusão do íon de lítio em P-Si 2 C 6 também pode ser muito rápido.
O desempenho da taxa de uma bateria está fortemente relacionado ao estado de carga (SOC), ou seja, a difusão de íons de lítio depende da concentração de adsorção de lítio. A fim de avaliar a dinâmica de difusão do íon de lítio em diferentes SOC, duas concentrações extremas são consideradas, a saber:íon de lítio diluído e vacância de lítio diluído. Para simulação da difusão diluída de íons de lítio, um íon de lítio é adsorvido em uma supercélula do P-Si 2 C 4 . Com base nas discussões acima na seção "Adsorção de Li no Penta-siligrafeno", o íon de Li prefere ficar no local B1 quando a concentração de Li é baixa, como mostrado na Fig. 7. Considerando a simetria da estrutura, apenas uma via de migração de Li ( denotado como Caminho-1 na Fig. 7) pode ser encontrado e o Caminho-1 forma uma rede de difusão de Li 2D completa no P-Si 2 C 4 superfície. A barreira de energia de migração de íons de lítio diluída em P-Si 2 C 4 ao longo da via otimizada do método NEB é de cerca de 0,117 eV, que é menor do que em P-C 6 (0,17 eV, Path-II) [21] e g-Si 2 C 4 (0,548 eV) [13]. When the SOC becomes 50%, namely, the material is discharged into the state of Li2 Si 2 C4 , all Li ions still occupy the B1 sites (see Fig. 3b) and thus the Li-ion diffusion pathway is the same as the case of dilute Li ion. These results show that Li-ion diffusion can be very fast at the beginning half of the discharge process.

The dilute Li-ion migration pathway and the corresponding energy profile along the pathway on P-Si2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The red arrows illustrate the two-dimensional diffusion networks
For the case of dilute Li vacancy, we remove one Li ion from the fully lithiated state Li4 Si 2 C4 and create one dilute Li vacancy in the supercell. As discussed above, Li ion prefers to occupy the H site when the Li concentration is high (see Fig. 3c). Therefore, three different Li vacancy migration pathways are considered, as shown in Fig. 8a. Path-1 refers to the Li vacancy migration from the H site to its neighboring H site across the top of a Si atom (across T site). Path-2 corresponds to the pathway across the top of the middle point of the C2–C2 dimer at the top layer (across B2 site). Path-3 is the pathway along the C2–C2 dimer at the down-layer (across B1 site). The energy profiles along the optimized pathways are given in Fig. 8b. As is seen, the Li-ion migration energy barriers along these pathways are very low, particularly for Path-3 (0.052 eV). The energy profiles along Path-1 and Path-2 are slightly asymmetric, because of the large relaxation of the Li ions when one Li vacancy is created. The extremely low energy barrier along Path-3 is reasonable, since Path-3 crosses B1 site (energetically most favorable adsorption site). However, Path-3 alone is not able to form a complete Li diffusion network on the surface of the P-Si2 C4 . Therefore, Path-1 or Path-2 must take part in the diffusion process, and the overall energy barrier for dilute Li vacancy migration is 0.155 eV or 0.165 eV. Although higher than dilute Li-ion migration (0.117 eV), the energy barrier for dilute Li vacancy migration is also very small compared with that for P-C6 (0.25 eV, Path-II’) [21] and g-Si2 C4 (0.233 eV) [13]. As the Li migration energy barriers in P-Si2 C4 are always lower than those in P-C6 and g-Si2 C4 (both dilute Li ion and dilute Li vacancy), it is expected that the rate performance of the P-Si2 C4 is the best one among the three similar anode candidates.

a Dilute Li vacancy migration pathways and b the corresponding energy profiles on fully lithiated P-Li4 Si 2 C4 . The gray (middle sized), golden, and purple spheres are C, Si, and Li atoms, respectively. The large green sphere represents the dilute Li vacancy. The thick/thin arrows indicate fast/slow migration pathways which form the two-dimensional diffusion networks
Conclusão
In summary, based on first principles calculations, we predicted that 2D pentagonal Si/C compound P-Si2 C4 can be potentially used as anode materials for LIBs. Phonon dispersion data confirmed the dynamic stability of the P-Si2 C4 structure at ground state, while AIMD simulation shows that the structure of the P-Si2 C4 can be stable at temperatures as high as 2000 K. The unique 2D buckled pentagonal structure promotes special empty C-2p z states that facilitate Li adsorption on the surface of the P-Si2 C4 , which offers a gravimetric Li storage capacity of 1028.7 mAhg −1 . The calculated dilute Li-ion/Li vacancy migration energy barriers show that Li-ion diffusion on the surface of the P-Si2 C4 can be faster than both the pentagonal graphene (P-C6 ) and the honeycomb-structured siligraphene. The metallic electronic structure of the lithiated P-Lix Si 2 C4 ensures good electronic conductivity of the material as electrodes. These advantages are crucial features to the P-Si2 C4 as a promising anode material for LIBs. In summary, our first principles study offers a novel strategy to design high-performance Si/C complexity for the application in LIBs.
Disponibilidade de dados e materiais
Todos os dados gerados ou analisados durante este estudo estão incluídos neste artigo publicado.
Abreviações
- 2D:
-
Bidimensional
- AIMD:
-
Ab initio molecule dynamics
- bcc :
-
Body-centered face
- CINEB:
-
Climbing image nudged elastic band
- DFT:
-
Teoria da densidade funcional
- DOS:
-
Densidade de estados
- EVs:
-
Electric vehicles
- GGA:
-
General gradient approximation
- HSE06:
-
Heyd-Scuseria-Erznerhof
- LIBs:
-
Li-ion batteries
- PAW:
-
Onda aumentada do projetor
- PBE:
-
Perdew-Burke-Ernzerhof
- SOC:
-
State of charge
- VASP:
-
Pacote de simulação Ab initio de Viena
- vdW:
-
van der Waals
Transporte aprimorado de elétrons da interface do cátodo PF-NR2 por nanopartículas de ouro
Estabilidade da forma de nanoplacas metálicas:um estudo de dinâmica molecular
Nanomateriais
- Scalmalloy:O mais recente material de alto desempenho para impressão 3D em metal
- Nanocristais de estanho para bateria futura
- Síntese fácil de nanopartículas de SiO2 @ C ancoradas em MWNT como materiais de ânodo de alto desempenho para baterias de íon-lítio
- Composto de MoS2 / Acetileno com poucas camadas como um ânodo eficiente para baterias de íon-lítio
- Preparação de micrromateriais híbridos MnO2 revestidos com PPy e seu desempenho cíclico aprimorado como ânodo para baterias de íon-lítio
- Efeito de diferentes ligantes no desempenho eletroquímico do ânodo de óxido de metal para baterias de íon-lítio
- Na4Mn9O18 / Composto de Nanotubo de Carbono como um Material de Alto Desempenho Eletroquímico para Baterias Aquosas de Íons de Sódio
- Composto de Si / Grafeno incorporado fabricado por redução térmica de magnésio como material de ânodo para baterias de íon-lítio
- Nanofolhas V6O13 interconectadas 3D cultivadas em têxteis carbonizados por meio de um processo hidrotérmico assistido por sementes como cátodos flexíveis de alto desempenho para baterias de íon-l…
- Um ânodo de filme Fe2O3 nanocristalino preparado por deposição de laser pulsado para baterias de íon-lítio